Adding Bam/Cram Tracks
In the web version of Persephone, the users can import BAM or CRAM tracks with NGS read alignments via drag & drop. Such tracks will be saved in the user's storage and will not be public. An alternative to this is to add the BAM/CRAM tracks to the database using the command
add bam -c <control-file> [-t|-v]
When the tracks are added using PersephoneShell, they will be shown to all users. Behind the scene, the BAM/CRAM files can be referenced as local files or by a URL. You can control where the alignment data from the file is stored. One option is to use the Persephone's file storage. It is similar to the way the genomic sequences are stored in case of MariaDb - the compressed sequence files are saved outside the database in the file system of the server, and the metadata for the records are saved in the database. The other option is to keep referencing the BAM/CRAM file by a URL. The Persephone client will read the index from the database and pull the alignment data from the remote file via a URL.
When loading the BAM/CRAM data with PersephoneShell, consider three parameters:
1. either read the BAM/CRAM file via URL or use a local copy;
2. in case of remote file, ask to copy it to the local storage (SaveToStorage=true) or keep it at the remote URL;
3. either create the detailed histogram (CreateDetailedHistogram=true) or not.
The simplest and fastest way of loading the data is to keep the BAM/CRAM data at the remote location and download only the original index provided as a .bai or .csi or .crai file. Such an index is usually small and, when the data files are located at a remote URL, can be easily transferred to the server without much of a delay. The remote file will not occupy any extra space on disk, but reading it over a slow network can degrade the performance of displaying the data in Persephone.
One optional step in loading the BAM/CRAM file is building a detailed index with the histogram. This is applicable to the local or remote files.
Please take a look at the picture below:
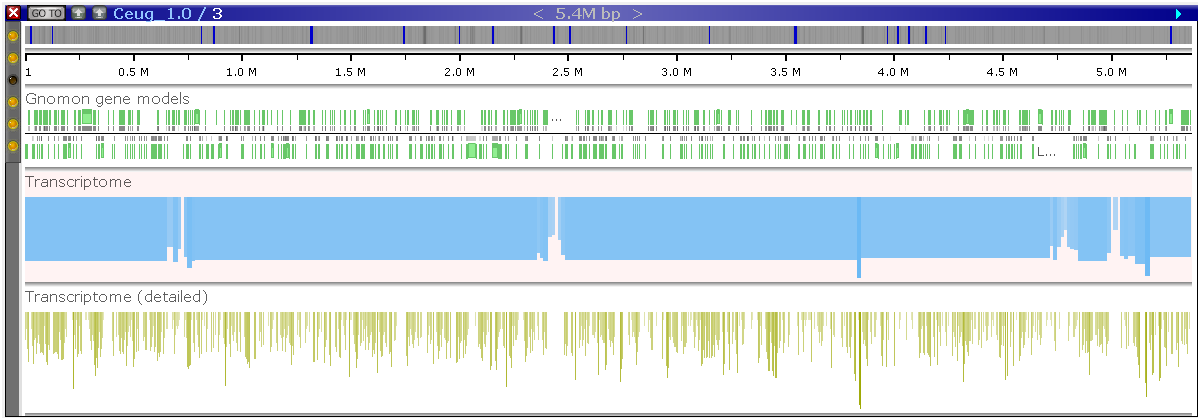
The two BAM tracks in the picture have been loaded using the same source file. At this resolution, Persephone does not show the individual reads but tries to indicate the density of the alignments by reading the index. The top (blue) track is drawn from the original .bai index. The height of the histogram is based on the size of data blocks in the index. It gives a very rough picture of the read distribution.
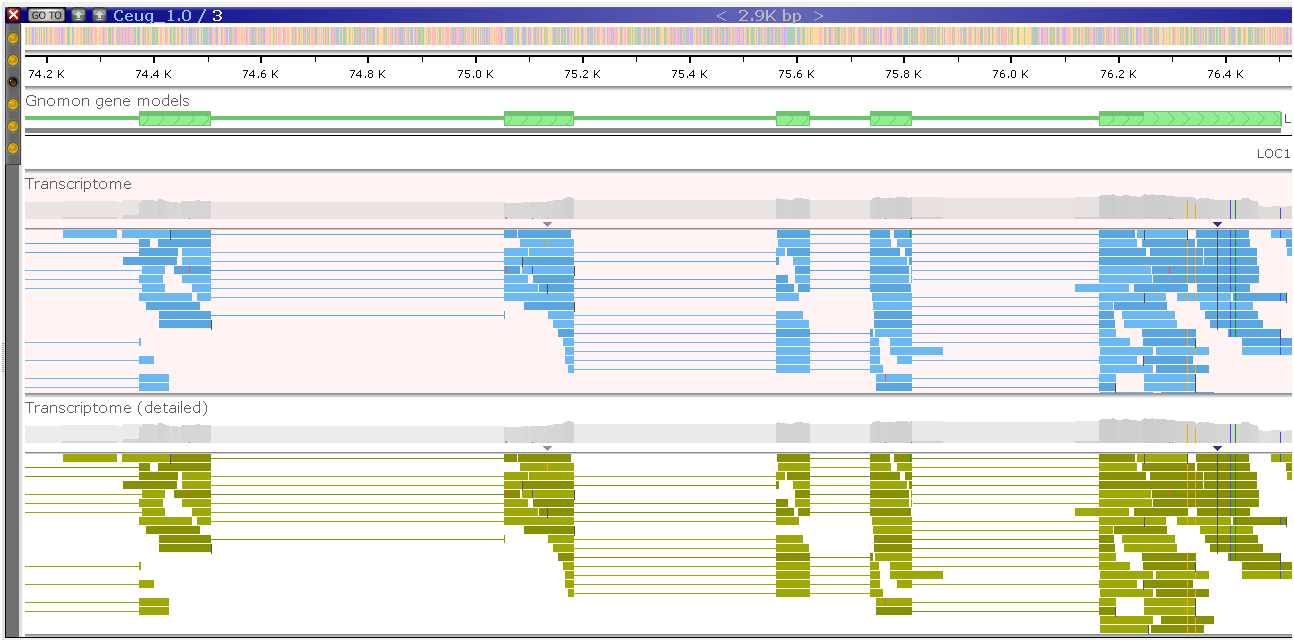
As another option, you can request to build a detailed histogram (CreateDetailedHistogram=true). It can be generated for a local or a remote file and will provide a much more detailed picture (the bottom track). Please note that once you zoom in deep enough, you will see the detailed read alignment for both versions of the track:
As a summary, here is a guide for the combination of the two variables: (1) local file vs. a URL and (2) use the file storage or not:
|
URL or local file |
Use file storage |
Pro |
Contra |
|
URL |
Yes |
Fast retrieval of track data from the local storage, data availability is guaranteed even if the original data source is deleted |
Uses extra disk space |
|
local file |
Yes |
Fast retrieval of track data from the local storage, data availability is guaranteed even if the original data source is deleted |
Uses extra disk space |
|
URL |
No |
Reasonably fast retrieval of track data from the URL, depending on the network conditions at the remote resource. Does not use much of the disk space |
Risk that the original file is removed. Can be slow if hosted on a weak resource. |
|
local file |
No |
Fast retrieval of track data from the original file, speed is similar to reading the data from the file storage. |
Risk that the original file is removed |
Note, the files in the storage take approximately the same disk space as the original file.
The control INI file
[ProcessRun]
; Run description: if specified, a custom description will be used,
; otherwise, "Loaded BAM for <MapSet AccessionNo> from <Sources>." will be used.
;RunDescription="Test adding BAM"
[MapSet]
; Either MapSetId or MapSetPath is required.
; MapSetId: id of a target map set.
;MapSetId=247582504
; MapSetPath: path of a target map set.
MapSetPath="/Coffee/Coffea eugenioides/Ceug_1.0"
[Bam]
; Source of index file, .bai or .csi file
;IndexSource="$DATA/coffee/coffea_eug.bam.bai"
IndexSource=http://www2.persephonesoft.com/coffea_eug.bam.bai
; Source: URL/filepath where BAM or CRAM file is
;Source="$DATA/coffee/coffea_eug.bam"
Source=http://www2.persephonesoft.com/coffea_eug.bam
; SaveToStorage: if 'true', BAM file is downloaded or copied from its original location and saved as a set of files in Storage.
;Set it to true if there is a risk that the original file will be removed or its network access is slow
SaveToStorage=true
; CreateDetailedHistogram: if 'true', a detailed histogram is created by reading through the entire BAM/CRAM file.
; The downside of this is a much longer processing time. 'false' by default.
;CreateDetailedHistogram=true
; TrackName: name of the track
TrackName="Transcriptome from a URL"
; TrackDescription: track description, optional
TrackDescription="Coffea eugenioides transcriptome from NCBI: SRR1768422.fastq"
; TrackColor: {NamedColor|HTML hex code|R,G,B}, optional
TrackColor=Green
; IsShownFirst: if false, the track will not be shown by default when the map is opened for the first time. true by default
IsShownFirst=false
[MapMapping]
; Normally, the map names in the BAM/CRAM files should correspond (case sensitive) to the map names in the database.
; In such case, no map name matching is necessary and this section can be empty.
; If map names in file are different from those in DB, you should map each MAP_NAME in the file to a MAP_NAME in DB.
; manual mapping below:
;MAP_NAME_IN_FILE=MAP_NAME or MAP_ID or ACCESSION_NO in DB
;Chr1=Chr.1
;Chr2=Chr.2
;Chr3=Chr.3
; Check 'printmapping' command that may help generating the name mapping tables.
; Alternatively, use MapIdentifiedBy
; MapsIdentifiedBy: if all maps in the file instead of the map name are identified by their alternative IDs like MAP_ID, ACCESSION_NO or GENOME_DNA_ID,
; provide the mapping with just one line using either MapName, MapId, AccessionNo or GenomeDnaId, for example:
; MapsIdentifiedBy: one of MapId, AccessionNo, GenomeDnaId, MapName (default)
;MapsIdentifiedBy=AccessionNo
; LoadListedMapsOnly: if true, only data for the maps listed in this section will be added.
; If false, PersephoneShell will still try to match names from the file to maps in the database
; using MAP_NAME, and if the map is not found, the data line will be skipped
;LoadListedMapsOnly=true
 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.