Adding Protein Alignments
Note
The track of this type is shown in the Windows version of Persephone only (which is discontinued). We plan to implement this feature in the web version.
A track with multiple aligned proteins can be helpful in annotating genomic regions, even if the gene models are not known yet. Such track can be produced by loading tblastn alignments of well characterized proteins (e.g., UniProt). Each graphical element in this track will represent a BLAST HSP (similar to a real-time BLAST track in Persephone).
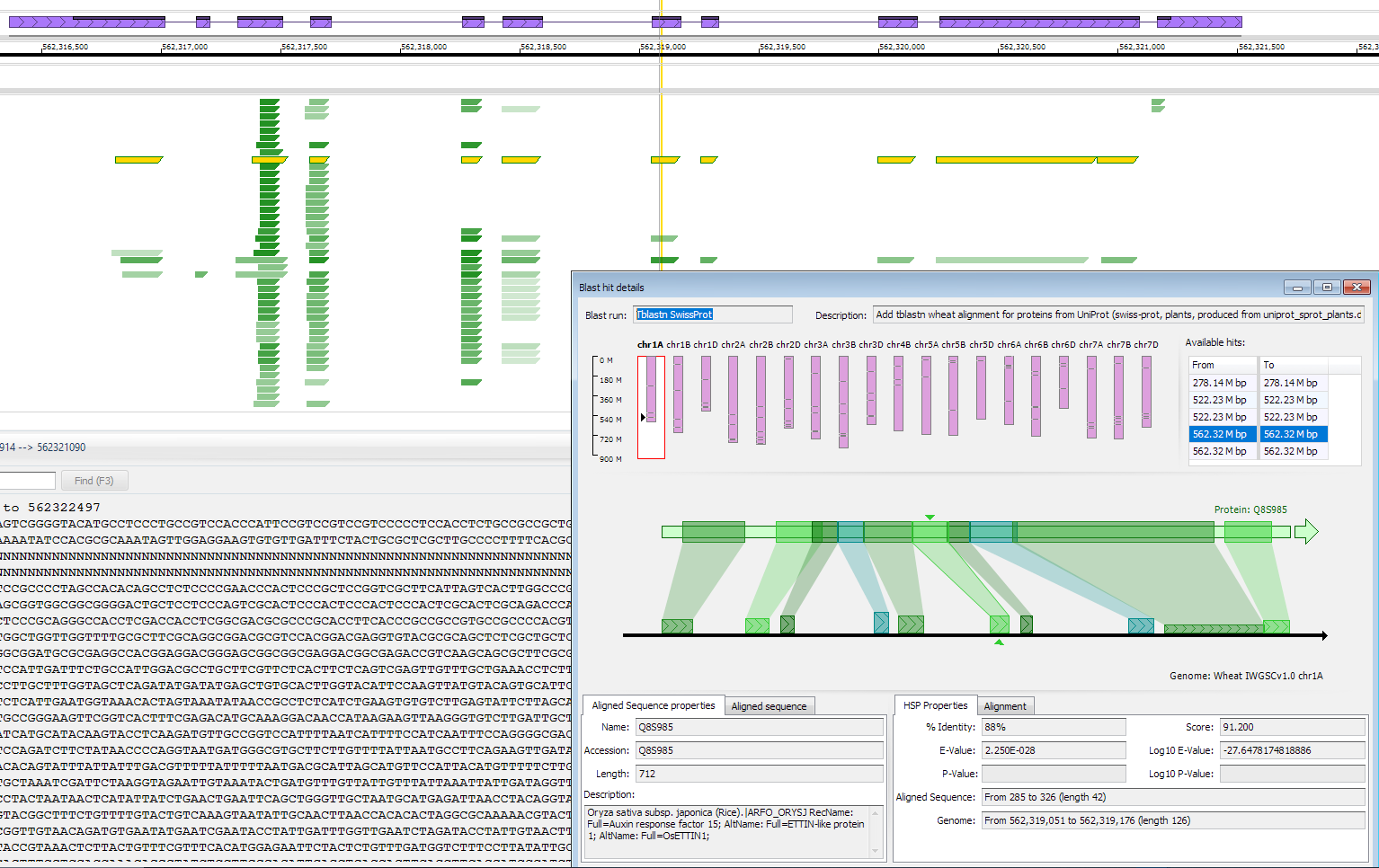
If the HSPs are compatible with each other and can form a chain, they will be highlighted together on mouse over. A click on any of the HPS elements will display the distribution of the hits on the query and on the genomic sequence together with the description of the aligned protein and other details.
The command for adding the alignments is
add alignment <-c controlFile> [-v] [-d] [-t | -f]
The alignments are typically available as the result of tblastn run stored as a tab-delimited file. Please check one of the sample files available in the Samples/Alignment sub-directory of PersephoneShell (the comments in the INI file are important):
; Sample file for loading tblastn results between a UniProt subset of plant proteins and cacao genome
[ProcessRun]
; RunDescription: (required). Describ the details of the alignments.
RunDescription="Add tblastn alignment for proteins from UniProt (advanced filter:Taxonomy=viridiplantae, Existence=evidence at protein level)"
; QuerySequenceType (required): type of query sequence {Nucleotide|Protein}.
; Use Nucleotide for blastn and Protein for tblastn
QuerySequenceType=Protein
; AlignedSequenceUrl: a base URL of the from 'http://www.server.com?seq_id=%s'.
; The '%s' will be replaced with the accession of the blast aligned sequence.
; This allows Persephone to retrieve the sequence at runtime. (Optional)
AlignedSequenceUrl="https://www.uniprot.org/uniprot/%s.fasta"
; AlignedSequenceView: the name of a table or view where the full sequences are stored.
; This view must have two columns: 'seq' and 'seq_accession'.
; Persephone will query this view at runtime in order to retrieve the sequence by its accession. (Optional)
;AlignedSequenceView=
[MapSet]
; Either MapSetId or MapSetPath is required.
; MapSetId: id of a target map set.
;MapSetId=200493023
; MapSetPath: path of a target map set.
MapSetPath="Theobroma cacao/Tcacao_CGDv1.1"
[Alignment]
; Output of tblastn. Additionally, the information about the proteins can be parsed from the FASTA file.
; Source (required): a file located locally or remotely accessible via URL.
Source=$DATA/uniprot/cocoa.blastout
; FASTA Source for the aligned sequences. Description and sequences can be parsed out of this file (Optional)
FastaSource=$DATA/uniprot/uniprotPlants.fasta
; LoadFastaSequence: The protein sequences from the FASTA file can be loaded into the database (Optional-not implemented)
LoadFastaSequence=false
; FastaDescriptionRegex: The regular expression used to extract the protein description from the FASTA header (Optional)
FastaDescriptionRegex=^.*?\|(.+)$
; FastaAccessionRegex: The regular expression used to extract the protein accession from the FASTA header (Optional).
; If the regex is not provided, the entire FASTA header will be stored as the protein sequence accession.
FastaAccessionRegex=^(.+?)\|
; Commit frequency: indicates how often the process commits records. Every N records.
CommitFrequency=1000
; FileType: {BlastTab(tabular BLAST output)|TeraBlast|Gff}
FileType=BlastTab
; Track name (required): name of track
TrackName="Tblastn UniProt"
; TrackDescription: track description
TrackDescription="tblastn alignment for proteins from UniProt (advanced filter:Taxonomy=viridiplantae, Existence=evidence at protein level).
TBLASTN command line:
tblastn -query uniprotPlants.fasta -db 29_N -evalue 1e-6 -threshold 999 -culling_limit 10 -outfmt \"6 qseqid sseqid pident length mismatch qstart qend sstart send evalue bitscore qseq sseq\" -matrix PAM70 -gapextend 2 -gapopen 8 -num_threads 6"
; QueryNameDelimiter: specify one among Colon(:), Comma(,), Period(.), Hyphen(-), SemiColon(;), Slash(/), Tab(\t), Space( ) and VerticalBar(|)
QueryNameDelimiter=VerticalBar
; Query name index (required): 0-based
QueryNameIndex=1
; ParentGroupName: the new track will be placed under a parent node with this name.
; To reduce the number of track nodes on the top level, group the tracks of similar type.
;ParentGroupName=tblastn tracks
; IsShownFirst: if false, the track will not be shown by default when the map is opened for the first time
IsShownFirst=false
[BlastTab]
; Delimiters: specify one or more, Colon(:), Comma(,), Period(.), Hyphen(-), SemiColon(;), Slash(/), Tab(\t), VerticalBar(|), Space( )
Delimiters=Tab
; a typcal TBLASTN output:
;qseqid sseqid pident length mismatch qstart qend sstart send evalue bitscore qseq sseq
;Q6V4H0 1 69.006 171 53 72 242 4715 5227 2.80e-94 198 HEIVG... HEIVG...
; List of fields in the tab-delimited file:
;QueryName - qseqid
;SubjectName - sseqid
;PercentIdentity - pident
;QueryStart - qstart
;QueryEnd - qend
;SubjectStart - sstart
;SubjectEnd - send
;EValue - evalue
;BitScore - bitscore
;QueryAlign - qseq
;SubjectAlign - sseq
;Ignore - the field will be ignored
; Fields: the meaning and the order of values in each TBLASTN output line
Fields=QueryName,SubjectName,PercentIdentity,Ignore,Ignore,QueryStart,QueryEnd,SubjectStart,SubjectEnd,EValue,BitScore,QueryAlign,SubjectAlign
; Extract the protein query name from the column with the query (optional)
;QueryNameRegex=^(.+?)\|
[MapMapping]
; If the map names in the BLAST output files correspond (case sensitive) to the map names in the database,
; no map name matching is necessary and this section can be empty.
; If the map names in file are different from those in DB, you should map each MAP_NAME in the file
; to a MAP_ID or an ACCESION_NO or MAP_NAME in DB.
; manual mapping below:
;MAP_NAME_IN_FILE=MAP_NAME or MAP_ID or ACCESSION_NO in DB
;Chr1=Chr.1
;Chr2=Chr.2
;Chr3=Chr.3
; Check 'printmapping' command that may help generating the name mapping tables.
; Alternatively, use MapsIdentifiedBy to show what system of sequence identifiers have been used.
; MapsIdentifiedBy: one of MapId, AccessionNo, GenomeDnaId, MapName (default)
; If all maps in the file instead of the MAP_NAME are identified by their alternative IDs
; like MAP_ID, ACCESSION_NO or GENOME_DNA_ID,
; provide the mapping with just one-line instruction using either MapName, MapId, AccessionNo or GenomeDnaId.
; It is typical to use BLAST index files prepared for Persephone in offline analysis.
; Note that the genomic sequences in such files are identified by GENOME_DNA_ID (an sequence identifier).
;MapsIdentifiedBy=AccessionNo
MapsIdentifiedBy=GenomeDnaId
; LoadListedMapsOnly: if true, only data for the maps listed in this section will be added.
; If false, PersephoneShell will still try to match names from the file to maps in the database
; using MAP_NAME, MAP_ID or ACCESSION_NO, and if the map is not found, the annotation line will be skipped
;LoadListedMapsOnly=true
[DbSequences]
; The ID columns below are used to add alignments (Oracle only)
;TABLE_NAME.COLUMN_NAME=SEQUENCE_NAME
;BLAST_RUN.BLAST_RUN_ID=ID_SEQ
;BLAST_ALIGNED_SEQ.BLAST_ALIGNED_SEQ_ID=ID_SEQ
;BLAST_HIT.BLAST_HIT_ID=ID_SEQ
;BLAST_HSP.BLAST_HSP_ID=ID_SEQ
;TRACK.TRACK_ID=ID_SEQ
;TRACK_TYPE.TRACK_TYPE_ID=ID_SEQ
;DESCRIPTION.DESCR_ID=ID_SEQ
Having just the tblastn output may not be enough - besides the coordinates of the HSPs the users would probably want to see the functional annotation and the source organism of the aligned proteins. All this information may be available in the FASTA file with the protein sequences used for the tblastn search.
To upload this information about the aligned proteins use the instructions FastaSource, FastaDescriptionRegex and FastaAccessionRegex. Having the protein accession will allow linking the query protein name in the tblastn output and the description read from the FASTA file.
If the sequences can be downloaded by a URL, provide the URL template in AlignedSequenceUrl, the actual URL will be formed by replacing '%s' with the protein accession value. If the protein sequence is available somewhere in the database, fill the value of AlignedSequenceView. It should point to the database view that has just two columns: 'seq_accession' (equal to the protein accession) and 'seq'.
In case tblastn is used to produce the alignments, make sure that the following output format is used: -outfmt "6 qseqid sseqid pident length mismatch qstart qend sstart send evalue bitscore qseq sseq". The Fields instruction shown in the example expects the output columns in the same order as listed after -outfmt parameter of tblastn.
 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.