Adding Genomic Sequences
Maps, based on genomic sequences, are loaded in a form of a "physical" (sequence-based) map set. The INI control file should specify the location of the sequence files, the name and other attributes of the map set, its placement in the map set tree, parameters of parsing FASTA definition lines to extract the map name, accession, etc. The source FASTA files may contain a mixture of chromosomes (Persephone will show them first) and scaffolds. The INI file can specify criteria on how to separate chromosomes from the rest of the sequences.
The sequences can be added to:
Creating a map set
Each loading process has a ProcessRun record that captures the logistics of operation: the time stamp, the operator's user name, the type of data loaded, etc. If you omit RunDescription, it will be generated using AccessionNo and Sources. Please read the comments in the sample control files, they are an essential part of the documentation. The sample files for loading sequences are stored in ./Samples/Sequence sub-folder of the PersephoneShell package.
[ProcessRun]
; RunDescription: if specified, a custom description will be used,
; otherwise, "Added sequences for {MapSet Accession No.} from {Sources}." will be used.
RunDescription="Load sequences for rice IR64 ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/009/914/875/GCA_009914875.1_OsIR64RS1"
Normally, the sequence maps are added to a new map set for which we should provide the name, accession and an organism (please note, the corresponding Organism record should already exist in the database).
[MapSet]
;------------------------------------------------------------------------------------------------
; 1. Using existing MapSet
;------------------------------------------------------------------------------------------------
; If either MapSetId or MapSetPath is specified, it adds sequences to the existing MapSet.
; otherwise, a new MapSet should be specified below.
; MapSetId: id of an existing map set
;MapSetId=270000441
; MapSetPath: path of an existing map set.
;MapSetPath="Glycine max/Wm82.a2.v1"
;------------------------------------------------------------------------------------------------
; 2. Adding new MapSet
;------------------------------------------------------------------------------------------------
; Organism ID (required): organism ID should exist.
OrganismId=4530
; Display name (required): a name shown in MapSetTree. Usually a assembly build name.
DisplayName="Rice IR64"
; Description: by default, organism name + display name. Note that the internal quotation marks should be escaped with back slash '\'
Description="<i>Oryza sativa</i> from NCBI.
IR64 (IRRI)
ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/009/914/875/GCA_009914875.1_OsIR64RS1/GCA_009914875.1_OsIR64RS1_genomic.fna.gz
Submitter: University of Arizona.<b>Twelve Platinum-Standard Reference Genomes Sequences (PSRefSeq) that complete the full range of genetic diversity of Asian rice.</b>
doi: https://doi.org/10.1101/2019.12.29.888347
NCBI Accession: <a href=\"https://www.ncbi.nlm.nih.gov/assembly/GCA_009914875.1\">GCA_009914875.1</a>"
; AccessionNo (required): accession of the genome build. See http://ncbi.nlm.nih.gov/genome
AccessionNo="GCA_009914875.1"
; Source ID: database or institution that the MapSet/sequence originate
SourceId="UA"
Note
A common mistake, that is sometimes difficult to catch, is leaving un-escaped quotation marks in the middle of the description text. Please remember that multi-line string values should be always enclosed in quotation marks. If you have quotes in the middle of your text (such as HTML hyperlinks), the quotation marks should be escaped by back slash like \". (See the example above in the code). Without escaping, any quotation mark will be treated as a signal to start or to end the string, and this may cause parsing problems.
Placement in the map set tree
The map set is placed in the map set tree. This can be done by specifying ParentNodeId or RootNodeName.
If you are going to reference an existing parent node in the map set tree by its ID, you will need to find its NodeId by running the command:
PS> list mapsettree -l
Arabidopsis thaliana (NodeId:1)
Physical:TAIR10 (NodeId:2, MapSetId:1)
Oryza sativa japonica (NodeId:3)
Physical:MSU_osa1r7 (NodeId:4, MapSetId:2)
Oryza sativa indica (NodeId:5)
Physical:ASM465v1 (NodeId:14, MapSetId:13)
Sorghum bicolor (NodeId:18)
Physical:Sorghum v.3.1 (NodeId:20, MapSetId:17)
Genetic:BTx623-IS320C (NodeId:21, MapSetId:18)
Use NodeId (=3) as ParentNodeId, and the newly created map set will be placed as a child of Oryza sativa japonica node:
[MapSetTree]
;------------------------------------------------------------------------------------------------
; 1. Adding new MapSetTree node to a parent node
;------------------------------------------------------------------------------------------------
; Parent node ID: if specified, the MapSet with the new sequences will be placed under this parent node as a child.
ParentNodeId=3
;------------------------------------------------------------------------------------------------
; 2. Adding new MapSetTree node under a new root node
;------------------------------------------------------------------------------------------------
; RootNodeName: usually an organism name. A longer path consisting of more than one node can be specified here. The nodes will be created starting with the root node.
;RootNodeName="Plants/Glycine max"
; OrderNo: placement of map set under its parent node in the map set tree. If not specified, OrderNo will be automatically incremented for each new map set.
; If you want to order all map sets under a parent node alphabetically, set OrderNo to the same fixed value. All map sets will get the same value and will be sorted alphabetically by name.
;OrderNo=0
Alternatively, you can specify the new position by using RootNodeName. This value can include more than one node starting with the root node. All missing nodes will be created. The leading slash is not required.
RootNodeName="Plants/Glycine (genus)/Glycine soja"
Position of the map set name listed under a parent node is controlled by OrderNo. By default, the new map set is assigned the next OrderNo. This guarantees that the map sets will be listed in the order they have been loaded. In case OrderNo values for several map sets are the same, they will be sorted alphabetically. If you prefer alphabetical sorting for all map sets under a given parent node, give them the same OrderNo, for example:
OrderNo=0
Tip
It is recommended to use the textual RootNodeName instead of the numeric ParentNodeId. The node names are much more portable, which allows reusing and sharing the INI files between database instances.
Sequence processing
The diagram below should help to overview the logic in loading and separating the sequences.
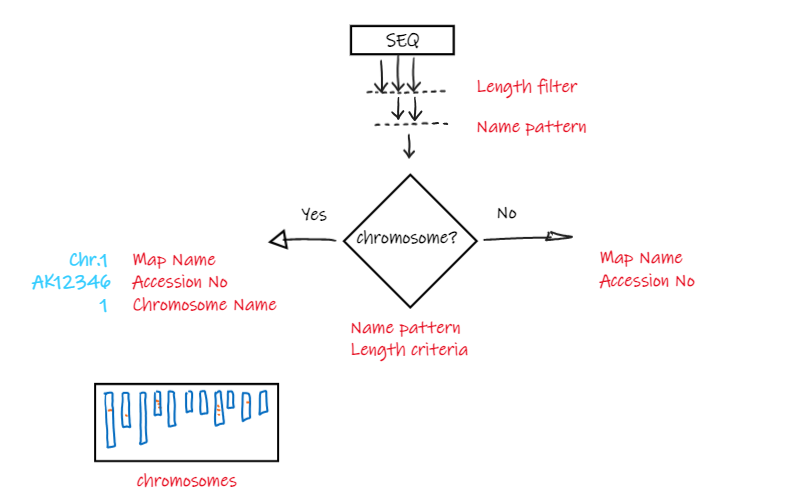
Genomic sequences can first be filtered by length (LengthFilter) or by regular expressions applied to the full FASTA header (ExcludedHeadersRegEx, IncludedHeadersRegEx). When any of these filters are enabled, only sequences that satisfy the criteria are loaded and processed further.
Next, the sequences are separated into chromosomes and non‑chromosomal entries. This distinction is needed because chromosome sequences are used to generate graphical genome representations, typically shown in search results. For this reason, chromosome names should be as short as possible, since many labels must fit into a single visualization. The chromosomes have priority over other sequences and are always shown first in the list of maps.
Finally, the system extracts the Map Name, Accession Number, and Chromosome Name from each FASTA header. The Map Name is a user‑friendly identifier displayed in the interface, while the Accession Number can be handy when gene annotations in GFF files reference sequences by accession (typical for NCBI files). If you want to preserve the original FASTA header, it can be saved into the map's description.
Important
Persephone displays the maps within a selected map set according to a defined ordering logic. Chromosomes always appear first, at the top of the list, and they retain the same order in which they were loaded from the original file. All remaining sequences are listed after the chromosomes and are sorted in reverse order by length. Keep this behavior in mind when deciding which sequences should be designated as chromosomes.
A simple case
If the FASTA headers already contain short identifiers like Chr.1, Chr.2, etc. (use the command analyze fasta to list the headers), then the entire header can be used directly as the Name, Accession and Chromosome name. In this case, you do not need to split the header or provide a regular expression to extract individual components.
If the FASTA file contains chromosomes only, our task is simple - we load all the entries, without filtering and selecting chromosomes. The entire [Sequence] section would look like this:
[Sequence]
; Sources (required): FASTA file(s) of genomic DNA sequence located locally or remotely accessible via URL.
Sources=$DATA/file.fasta
; Commit frequency: indicates how often the process commits sequence in bp.
CommitFrequency=10000000
You may need to use the logic nominating some maps as chromosomes, and this is usually done by giving a length threshold. In the example below, all sequences longer than 1,000,000 bp will be called chromosomes and will represent the genome in its "bird-eye" view.
; ChromosomeCriteriaLength - a formula to separate chromosomes from the rest of the sequences. It can be an alternative
; to ChoromosomeCriteriaRegEx
; For example, ChromosomeCriteriaLength=">5000000" will store sequences longer than 5,000,000 bp as chromosomes
ChromosomeCriteriaLength=">1000000"
A more complex case
When the FASTA sequence have complex headers, we will need to be more creative.
Suppose, we have a FASTA definition line like this:
>CM020876.1 Oryza sativa cultivar IR64 (IRRI) chromosome 1, whole genome shotgun sequence
We want the map name to be 1 and accession to be CM020876.1. To parse it out of the definition line, we first split the line by delimiter (Space) and then take the eighth field (0-based, index=7) for the map name. To get rid of the comma, we use the MapNameRegEx regular expression:
[Sequence]
; Sources (required): FASTA file(s) of genomic DNA sequence located locally or remotely accessible via URL.
Sources=$DATA/rice12/GCA_009914875.1_OsIR64RS1_genomic.fna.gz
; Commit frequency: indicates how often the process commits sequence in bp.
CommitFrequency=10000000
; SkipExistingSequences: When adding sequences to an existing map set and this flag is true, duplicated sequences will be automatically skipped.
SkipExistingSequences=true
; ExcludedHeadersRegEx: exclude sequences that match this regular expression
;ExcludedHeadersRegEx="chloroplast"
; IncludedHeadersRegEx: include only sequences that match this regular expression. Note, ExcludedHeadersRegEx, if specified, is applied first
;IncludedHeadersRegEx="complete"
; The FASTA header can be partitioned by splitting it by Delimiter or by applying regular expressions.
; Delimiter: specify one among Colon(:), Comma(,), Period(.), Hyphen(-), SemiColon(;), Slash(/), Tab(\t), Space( ) and VerticalBar(|)
; Comment this line out if the FASTA header will not be partitioned
Delimiter=Space
; Map name index. Tells which field between the delimiters corresponds to the map name. 0-based. If delimiter is not used, MapNameIndex is ignored,
; the entire FASTA header is saved as the map name.
MapNameIndex=7
; If the part between delimiters is too long to be stored as the map name, it can be parsed and the map name can be extracted by using this regular expression
; If MapNameIndex is not provided, MapNameRegEx is applied to the whole FASTA header
MapNameRegEx="(\d+),"
; ReplaceSpacesWith: Provide symbols that will be used to replace spaces in the map name. Some third-party tools are confused if the map name contains a space.
;ReplaceSpacesWith=_
; Map accession index. Tells which part between delimiters corresponds to map accession. 0-based. If delimiter is not used, MapAccessionIndex is ignored,
; the entire FASTA header is saved as the map accession.
MapAccessionIndex=0
; If the part between delimiters is too long to be stored as the map accession, it can be parsed and the map accession can be extracted by using this regular expression
; If MapAccessionIndex is not provided, MapAccessionRegEx is applied to the whole FASTA header
;MapAccessionRegEx=".*chromosome (..).*"
; Map description index
;MapDescriptionIndex=0
; MapDescriptionRegEx: regular expression to extract the map description from FASTA header or, if MapDescriptionIndex is provided,
; from the extracted field at that index
;MapDescriptionRegEx=(.*)
; MapDescriptionExtra: extra text appended to the description that can potentially embed map name ($MAPNAME) or accession ($MAPACCESSION) to form a hyperlink.
;MapDescriptionExtra=<a href=\"https://www.ncbi.nlm.nih.gov/nuccore/$MAPNAME\" target=\"_blank\">NCBI</a>
; Some sequences can be called chromosomes, some as scaffolds. Usually, the number of chromosomes is small and allows to show all of them as a representation of a genome.
; At least one criterion (ChromosomeCriteriaRegEx or ChromosomeCriteriaLength) should be used to enable parsing chromosome names.
; ChromosomeCriteriaRegEx - if the FASTA header matches the RegEx criteria, the entry will be called a chromosome
;ChromosomeCriteriaRegEx="chromosome"
; ChromosomeCriteriaLength - a formula to separate chromosomes from the rest of the sequences.
; For example, ChromosomeCriteriaLength=">5000000" will mark sequences longer than 5,000,000 bp as chromosomes
ChromosomeCriteriaLength=">5000000"
; If the sequence passes the criteria to be called a chromosome, the chromosome name will be read from the field with this index:
ChromosomeNameIndex=7
; ChromosomeNameRegEx - If the sequence passes the criteria to be called a chromosome, this regular expression can be used to extract the chromosome name.
; If ChromosomeNameIndex is specified, the regex is applied to the extracted field.
ChromosomeNameRegEx="(\d+),"
; Expected length index: if specified, the expected length of each sequence will be compared to the actual sequence length.
;ExpectedLengthIndex=5
; Length filter: used to include only sequences whose length satisfies the criteria. Use '>' or '<' to load sequences longer/shorter than the given number (e.g,"<1000000")
; if not specified, all the sequences in the source will be included.
LengthFilter=">1000000"
; ChromosomeReorder: force ordering chromosomes. By default (None), chromosomes are shown in the order they appear in the file.
; If ChromosomeReorder is Natural, chromosomes are ordered in natural order (chr1, chr2, ..., chr10, chr11, ...).
; If ChromosomeReorder is ByLength, chromosomes are ordered by length, longest to shortest.
;ChromosomeReorder=Natural
We use the same regular expression to extract the name of the chromosome.
ChromosomeNameRegEx="(\d+),"
In our case, we also load only long sequences (LengthFilter=">1000000"). Among those, the sequences longer than 5,000,000 will be called chromosomes (ChromosomeCriteriaLength=">5000000").
To nominate chromosomes you can also use the regular expressions - those that match the regex ChromosomeCriteriaRegEx will be stored as chromosomes. Please note that even if you provide the regular expression for extracting the chromosome name, the chromosomes will not be assigned unless you give one of the criteria for detecting chromosomes.
The order of chromosomes reported by Persephone follows the order of them in the original source file. If you want them to appear sorted, use the instruction ChromosomeReorder, which allows reordering them by "natural compare" (1,2..10) or by the sequence length. Please see the comments in the INI file.
Once the control file is ready, run PersephoneShell to test it first:
PS> add sequence -c pathToControlFile.ini -t
and, if all tests are successful, start the real loading:
PS> add sequence -c pathToControlFile.ini -v
It is worth noting that we have a switch -e to test and run the load in one command without giving control to any competing command waiting in a queue that may invalidate the test.
After all the sequences have been loaded, it is time to add annotation or other tracks.
Advanced case
Sometimes, the input FASTA file contains a mixture of sequences with headers in different patterns. Imagine a file with these headers of two types:
>NC_041021.1 Glycine soja cultivar W05 chromosome 20, ASM419377v2, whole genome shotgun sequence
>NW_021143571.1 Glycine soja cultivar W05 unplaced genomic scaffold, ASM419377v2 Super-Scaffold_110, whole genome shotgun sequence
We need to extract the map name (highlighted) from both formats using the same instruction.
To resolve this puzzle, let's use conditional regular expression, so that it will apply two different patterns for the two cases. First, we should stop using MapNameIndex, as we want to analyze the entire header. So, we have to comment it out. The logic of the map name extraction will be performed by the regular expression MapNameRegEx.
The regular expression can have two (or more) conditions and corresponding capture groups separated by a pipe (|) symbol. In case the first condition is true, the first group will work, otherwise, we will try the second condition, and, if it is successful, the second group will be used, and so on.
^NC.+(chromosome \d+)|, ASM419377v2 ([-\w]+)
We can distinguish the cases by looking for 'NC' at the beginning of the header. This condition will be written as:
^NC.+
So, if the header meets the first condition, we will use the first capture group:
(chromosome \d+)
Otherwise, the second condition and the capture group will be engaged:
, ASM419377v2 ([-\w]+)
This technique allows loading the chromosome and scaffold sequences in one shot. Remember that using the regular expression ChromosomeCriteriaRegEx or a length criteria ChromosomeCriteriaLength will tell which sequences should be marked as chromosomes.
Another example
Just for the sake of an exercise, let's imagine that we have a set of FASTA headers with two groups:
chromosomes:
NT_033777.3 Drosophila melanogaster chromosome 3R
NT_037436.4 Drosophila melanogaster chromosome 3L
NT_033778.4 Drosophila melanogaster chromosome 2R
NC_004354.4 Drosophila melanogaster chromosome X
and scaffolds:
NW_007931121.1 Drosophila melanogaster chromosome X; Y rDNA sequence
NW_007931076.1 Drosophila melanogaster chromosome 3CEN 3Cen_mapped_Scaffold_1_D1896_D1895 sequence
NW_007931108.1 Drosophila melanogaster chromosome Y Y_mapped_Scaffold_5_D1748_D1610 sequence
We want to extract the map names for the chromosome group and produce the regular expression that would capture "chromosome X" (highlighted) and not capture "chromosome X" from the group with the scaffolds. The distinguishing feature for these two cases is the end of line that follows the chromosome names in the first group. We will add the corresponding symbol $ at the end of the regular expression:
(chromosome .{1,2})$
Note also that we need to specify the number of characters (one or two) after "chromosome" to capture, otherwise, the regular expression chromosome .+$ would pull the entire text from "chromosome" till the end of the line.
One more example
Here is an example where we will need to
- filter the maps to be loaded excluding the entries that have 'Mitochondrion' in the header line
- cut out map names by using a regular expression to extract a part of the text
- cut out map accessions by first splitting the header at the 'TAB' symbol and using the first field (index=0)
- cut out chromosome name using regular expression
- identify chromosomes.
PS> analyze fasta Panicum_miliaceum.genome.fasta.gz
LENGTH HEADER
----------------------------------------------------------------------------------------------------------------------
67,993,370 GWHAAEZ00000001.1 Chromosome 1 Complete=F Circular=F OriSeqID=Chr01 Len=67993370
61,969,531 GWHAAEZ00000002.1 Chromosome 2 Complete=F Circular=F OriSeqID=Chr02 Len=61969531
58,135,681 GWHAAEZ00000003.1 Chromosome 3 Complete=F Circular=F OriSeqID=Chr03 Len=58135681
56,237,202 GWHAAEZ00000004.1 Chromosome 4 Complete=F Circular=F OriSeqID=Chr04 Len=56237202
53,988,371 GWHAAEZ00000005.1 Chromosome 5 Complete=F Circular=F OriSeqID=Chr05 Len=53988371
52,802,786 GWHAAEZ00000006.1 Chromosome 6 Complete=F Circular=F OriSeqID=Chr06 Len=52802786
50,975,384 GWHAAEZ00000007.1 Chromosome 7 Complete=F Circular=F OriSeqID=Chr07 Len=50975384
48,196,177 GWHAAEZ00000008.1 Chromosome 8 Complete=F Circular=F OriSeqID=Chr08 Len=48196177
45,128,071 GWHAAEZ00000009.1 Chromosome 9 Complete=F Circular=F OriSeqID=Chr09 Len=45128071
44,839,679 GWHAAEZ00000010.1 Chromosome 10 Complete=F Circular=F OriSeqID=Chr10 Len=44839679
42,762,355 GWHAAEZ00000011.1 Chromosome 11 Complete=F Circular=F OriSeqID=Chr11 Len=42762355
42,140,413 GWHAAEZ00000012.1 Chromosome 12 Complete=F Circular=F OriSeqID=Chr12 Len=42140413
40,727,217 GWHAAEZ00000013.1 Chromosome 13 Complete=F Circular=F OriSeqID=Chr13 Len=40727217
38,257,208 GWHAAEZ00000014.1 Chromosome 14 Complete=F Circular=F OriSeqID=Chr14 Len=38257208
34,254,202 GWHAAEZ00000015.1 Chromosome 15 Complete=F Circular=F OriSeqID=Chr15 Len=34254202
33,582,435 GWHAAEZ00000016.1 Chromosome 16 Complete=F Circular=F OriSeqID=Chr16 Len=33582435
33,245,457 GWHAAEZ00000017.1 Chromosome 17 Complete=F Circular=F OriSeqID=Chr17 Len=33245457
31,992,649 GWHAAEZ00000018.1 Chromosome 18 Complete=F Circular=F OriSeqID=Chr18 Len=31992649
28,187 GWHAAEZ00000019.1 OriSeqID=contig_1 Len=28187
11,092 GWHAAEZ00000020.1 OriSeqID=contig_2 Len=11092
5,295 GWHAAEZ00000021.1 OriSeqID=contig_3 Len=5295
17,333 GWHAAEZ00000022.1 OriSeqID=contig_4 Len=17333
14,918 GWHAAEZ00000023.1 OriSeqID=contig_5 Len=14918
12,203 GWHAAEZ00000024.1 OriSeqID=contig_6 Len=12203
7,783 GWHAAEZ00000025.1 Mitochondrion Complete=F Circular=F OriSeqID=contig_7 Len=7783
26,803 GWHAAEZ00000026.1 OriSeqID=contig_8 Len=26803
1,355 GWHAAEZ00000027.1 OriSeqID=contig_9 Len=1355
8,447 GWHAAEZ00000028.1 OriSeqID=contig_10 Len=8447
...
For some reason, there are several maps marked "Mitochondrion". We will exclude them from loading by using this instruction:
ExcludedHeadersRegEx="Mitochondrion"
We need to capture the map names like 'Chromosome 1' for the chromosomes and something like 'contig_1' for the other entries. To do that, we will use a composite regular expression:
MapNameRegEx=(Chromosome \d+)|OriSeqID=(.+)\t
Note that capture groups for the extracted text should be specified in the parenthesis: (Chromosome \d+) or (.+). If the first group fails, the next group is engaged, etc.
The accession number is the identifier at the beginning of the line. If we split the header by the 'TAB' character, we can use the first field (index=0) as the accession:
Delimiter=Tab
MapAccessionIndex=0
The chromosome names (not to be confused with map names for chromosomes) are short names that will appear in a graphical representation of a genome, that is why they should be as short as possible: many chromosomes are shown together. We will extract just the number 1 from the text 'Chromosome 1' by using the instructions:
ChromosomeNameIndex=1
ChromosomeNameRegEx=Chromosome (\d+)
First, we use the second field (index=1), then we extract the numeric characters that follow the word 'Chromosome'. To specify which maps will be nominated as chromosomes we will use the sequence length criteria:
ChromosomeCriteriaLength=">20000000"
The sequences longer than 20,000,000 bp will be called chromosomes.
Defining translation code
Some of the sequences in a map set can have a different translation code used to produce protein sequences from CDS. For example, the translation in chloroplasts uses the code 11. Specify this information in a separate section:
[TranslationCode]
; List map names and their corresponding translation codes. Default code is 1.
;Use wildcard '*' as a map name to assign the same code to all maps of the map set
chloroplast=11
Note that if the map name is set to the wildcard '*', all maps in the given map set will be assigned a common translation code.
Adding sequences to an existing map set
Sometimes it is necessary to add new sequences to an existing map set. You can reference the target map set either by its MapSetId (e.g., 55) or by its MapSetPath (e.g., /Homo sapiens/GRCh38). We strongly recommend using MapSetPath, because numeric IDs are not quite portable across database instances. If you later reuse the same INI file to populate a different database, using MapSetPath greatly increases the chance that the file will work without any modifications.
[MapSet]
;------------------------------------------------------------------------------------------------
; 1. Using existing MapSet
;------------------------------------------------------------------------------------------------
; If either MapSetId or MapSetPath is specified, it adds sequences to the existing MapSet.
; otherwise, a new MapSet should be specified below.
; MapSetId: id of an existing map set
;MapSetId=247582504
; MapSetPath: path of an existing map set.
MapSetPath=/Homo sapiens/pangenome/HG04187 paternal
When using the same source FASTA file with sequences to append the additional entries, use the construct
; SkipExistingSequences: When adding sequences to an existing map set and this flag is true, duplicated sequences
; will be automatically skipped.
SkipExistingSequences=true
Note:
The lines in the INI file needed to create a new map set will be ignored when the sequences are added to an existing map set.
 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.