Add Gene Annotation
After adding an organism along with its map sets, maps, and sequences using PersephoneShell, the next step is to incorporate gene annotation tracks. To add gene annotations, use the add command (see Add) to process a control file (see Control Files) containing instructions for loading the annotation data. This page shows the steps necessary to add gene models for Glycine max (soybean) genome to your Persephone database.
Please study the sample INI file and run the command add annotation that references this file. As usual, first, run the test (using the -t switch) and only if everything is correct, load the data to the database, preferably using -v for verbose output. As before, use auto-complete to speed up typing.
add annotation -c Samples/Annotation/Wm82.a4_phytozome-gff.ini -t
Sample file
Here is the sample file Wm82.a4_phytozome-gff.ini for our exercise:
[ProcessRun]
; Run description: if specified, a custom description will be used,
; otherwise, "Added annotation for {MapSet path} from {Sources}." will be used.
RunDescription="Load annotations for Wm82.a4.v1 from Phytozome"
[MapSet]
; Either MapSetId or MapSetPath is required.
; MapSetId: id of a target map set.
;MapSetId=247848026
; MapSetPath: path of a target map set. The command 'list path' will print paths for all map sets, with
; the latest ones shown at the end of the list
MapSetPath="/Glycine max/Wm82.a4.v1"
[Method]
; To add a new annotation method, specify a method name and a color to be shown in the annotation track.
; You don't need to provide this info if the method already exists in the database.
; Supported color name is available at http://www.flounder.com/csharp_color_table.htm
; If the method already exists, it will be updated. Otherwise, a new method will be added.
;Name={NamedColor|HTML hex code|R,G,B}
; Color examples: Red, #FF0000, or 255,0,0
;mRNA=203,128,147
Phytozome13=100,100,255
[Annotation]
; GFF specification at http://www.sequenceontology.org/gff3.shtml
; Sources (required): GFF or GTF file(s) for annotations, accessible locally or remotely via URL.
; It is important to keep the file name extension consistent with the file format.
; Files can be compressed with gzip.
; $DATA variable defined in psh.exe.config as DataDir can be used in the path.
Sources=$DATA/soy/Gmax_508_Wm82.a4.v1.gene_exons.gff3.gz
; MaskedParentTypes: parent GFF types (column 3) that together with their child records should be ignored
; This saves some memory during GFF parsing, especially if the GFF file contains many records of this type.
;MaskedParentTypes="biological_region","repeat_region"
; The GFF files are expected to have their records grouped by map. All records of a chromosome should come
; in one continuous block, followed by a block for another chromosome, etc. The records within the block can be unsorted (typical for Ensembl GFFs),
; the program will sort them in memory.
; SkipInconsistentRecords: indicates how to treat conflicting records. If SkipLine - we will try to create annotation without
; individual conflicting lines, SkipMRNA - the entire MRNA record will be skipped if at least one line of the record is conflicting others,
; None (by default) - nothing to skip. No case sensitivity.
;SkipInconsistentRecords=SkipMRNA
;AbuttingExons: exons that have an intron of size 0 between them. We can throw an Error (default), AddBoth or Merge such exons.
;AbuttingExons=AddBoth
; Commit frequency: indicates how often the process commits annotations to the database. Every N annotations.
CommitFrequency=1000
; Method (required): annotation method. If new, should be specified in the [Method] section.
Method=Phytozome13
; Column2: while parsing the gff file, only those lines will be analyzed that have the listed values in the gff column 2.
; Normally, this column shows the method or the source of the annotation.
; If the gff file has multiple sources in column 2, setting this filter will reduce the amount of data to be parsed and significantly improve the performance.
;Column2="maker"
; Track name (required): name of the track
TrackName="Gene models"
; TrackDescription: track description (note: the internal quotes should be escaped with back slash)
TrackDescription="Gene models predicted by Phytozome13
Downloaded from https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Gmax, file Gmax_508_Wm82.a4.v1.gene_exons.gff3.gz.
\"Best hits\" are defined as the top result returned from BLASTP alignment of this species proteome to the target
(A. thaliana, O. sativa, or C. reinhardtii listed above). This was run with blast+ 2.2.26 with parameters:
blastall -p blastp -F \"mS\" -b 1500 -v 1500 -e 0.001 -M BLOSUM45
and further filtered with an 1E-3 cutoff e-value."
; Type: GFF type (column 3) of annotation items. If not specified, both exon (SO:0000147) and CDS (SO:0000316) will be parsed.
; This line is typically left commented out
; The types of one-exon features can be defined here. To load multiple types, e.g., lncRNA, tRNA into one track,
; list them separated by comma. These items will be loaded as they are - no child records will be analyzed if ParentType is not specified.
;Type=lnc_RNA,tRNA
; Parent type (optional, typically commented out): the GFF type of parent items that groups annotation items,
; for protein-coding genes this is typically "mRNA". The child items, such as "exon" or "CDS", that have this common parent,
; will be grouped and form gene models. The types like "five_prime_UTR" or "three_prime_UTR" are not required but if present,
; will be checked for consistency with the exon and CDS records.
; If ParentType is omitted - the parent will be auto detected by taking direct parents of records of type 'exon' and 'CDS'.
; To load items other than 'exon' and 'CDS', Type and ParentType should be specified explicitly.
;ParentType="mRNA"
; Qualifier type: GFF type of parent items that contain qualifiers
QualifierType="mRNA"
; Qualifier attributes: qualifiers to be loaded from the GFF attribute (the last column of GFF).
; If QualifierTypes list more than one type, to disambiguate the qualifiers with identical names,
; use "fully-qualified" name which includes the particular QualifierType.
; For example, if "gene" and "mRNA" records requested in QualifierTypes both have qualifier "Name",
; to load only "Name" from the mRNA line, type "mRNA:Name"
; When running in the test mode, PSH will print the list of found attributes as one line, ready to be copied to this instruction
QualifierAttributes=Name,pacid,longest,ancestorIdentifier
; For the qualifiers that need renaming, use a more flexible way of loading qualifiers engaging QualifierAttributeKey instruction.
; The attribute key in GFF can be stored as a qualifier with a different name. For example, to disambiguate gene:Name and mRNA:Name,
; store them as two different qualifiers, "geneName" and "transcriptName" using this construct:
; QualifierAttributeKey.gene:Name=geneName
; QualifierAttributeKey.mRNA:Name=transcriptName
;QualifierAttributeKey.[Type:]AttributeKey=qualifierName
;QualifierAttributeKey.Note=Function
; The line above stores attribute "Note" as qualifier "Function"
; GroupNameQualifier: group splice isoforms by common qualifier. By default, the transcripts are grouped by location - all overlapping
; gene models will be assigned to the same group. This can be overridden by providing the qualifier name, and the models with the same
; value of the qualifier will belong to the same group. Note: parent_gene_id is an automatically added qualifier that captures the ID
; of the parent record
;GroupNameQualifier=parent_gene_id
; GroupByLocation and GroupNameQualifier are options to enable gene model grouping. This grouping is necessary for reducing the number of ortholog records.
; It is applied only to gene models that have CDS.
; GroupByLocation (default:true): set it to false (and don't provide GroupNameQualifier) if you want to disable grouping of the gene models.
; Note, without groups, orthologs cannot be created for such tracks.
;GroupByLocation=false
; New.: Create new qualifiers by extracting substrings from existing gff attributes using regular expression.
; The general form of the instruction is
;New.newQualifierName=[Type:]attributeKey,regex
;New.Pfam=gene:Dbxref,Pfam:(\w+)
;New.Prosite=gene:Dbxref,Prosite:(\w+)
;New.InterPro=gene:Dbxref,InterPro:(IPR\w+)
;New.GO=gene:Ontology_term,(GO:\w+)
; The line above creates a new qualifier 'GO' by extracting a substring from an attribute 'Ontology_term'.
; Note that the new qualifiers can also be created later, by using the command 'add annotation_qualifier'.
; ParentGroupName: the new track will be placed under a parent node with this name.
; To reduce the number of track nodes on the top level, group the tracks of similar type.
;ParentGroupName=gene models
; OrderNo: track's order number in the list of tracks for each map. By default, if it is not specified, it will be automatically assigned sequentially
; thus preserving the order of track loading. Sometimes, to force showing a track as the first one on a map, you can use negative number for OrderNo
;OrderNo=-1
; IsShownFirst: if false, the track will not be shown by default when the map is opened for the first time
;IsShownFirst=true
; IsSearchable: If true (default), the track data will be indexed for search. If false, the indexing will be skipped
;IsSearchable=false
; GroupMaxFeatures - All tracks in this loading procedure should have the number of features less than this value. If even one track has a higher feature count,
; the grouping will not be applied.
; The grouping is necessary for the tracks with gene models that will be used for finding orthologs. The ortholog pairs are recorded between the groups, not between
; individual genes. If a track contains features that have CDS but are not supposed to be grouped, avoid grouping by setting GroupMaxFeatures to a low number, e.g., 1000.
; This will result in the tracks in this batch not being grouped, and thus save you a lot of processing time.
; If GroupMaxFeatures is not used, the default value of 400000 will be used. To change the default value, modify the section <appSettings> in psh.exe.config:
; <appSettings><add key="MaxGenesOnTrackToGroup" value="300000" /></appsettings>.
;GroupMaxFeatures=100000
[QualifierLinks]
; Some qualifiers or their parts can be shown as hyper-links.
; Link the qualifier name-value to external sources.
; %s in the link will be replaced with the actual value of the qualifier
;QUALIFIER_NAME=PLACEHOLDER_URL[|regex]
;ID="http://rice.plantbiology.msu.edu/cgi-bin/gbrowse/rice/?name=%s"
; The line above would result in the qualifier "ID" shown as a hyper-link.
; For example, if ID="Os1g123", the URL would be "http://rice.plantbiology.msu.edu/cgi-bin/gbrowse/rice/?name=Os1g123"
Name="https://phytozome-next.jgi.doe.gov/report/transcript/Gmax_Wm82_a4_v1/%s"
; Optional: a qualifier link can be embedded into a longer text of the qualifier value. For example, 'Dbxref' can contain multiple identifiers, each of them
; can be used to construct a hyperlink. Use a regular expression to extract a substring that will be converted into the hyperlink inside the text. The regular expression
; should be appended using a pipe '|' symbol:
;Dbxref=https://www.ebi.ac.uk/interpro/entry/InterPro/%s/|Interpro:(IPR\d+)
; https://pfam.xfam.org/family/%s|(PF\d+)
; The command above contains two URL-regex pairs applied to the qualifier key 'Dbxref' separated by a new line.
; The first one instructs to analyze the qualifier value stored under key 'Dbxref', find substring that starts with 'Interpro:'
; and extract the part that has 'IPR' as the first letters followed by digits.
; The second one will extract substrings that start with PF and contain digits, and plug them into the placeholder %s of the URL that points to Pfam site.
; Later, you can use the PSH command 'add qualifier_link' to add more URLs and regular expressions.
[AnnotationSearches]
; Add qualifier name-value to search term {GeneName, GeneFunction}
; Some qualifiers can have a special meaning: they represent the gene name and gene function.
; This will help narrow down the text search specifically to the gene name or function in Persephone.
; This section will establish the distinction. The keyword GeneName tells which qualifier is designated as the gene name.
; Important: use the qualifier names here, which can be different from the GFF attribute names. For example, if you have introduced a qualifier "transcriptName" by
; renaming the GFF attribute mRNA:Name, then type GeneName=transcriptName.
; The instructions can refer to multiple qualifiers: GeneName=transcriptName,geneName
;SEARCH_TERM=QUALIFIER_NAME
GeneName="Name"
;GeneFunction=Description
[MapMapping]
; Normally, the map names in the GFF files should correspond (case sensitive) to the map names in the database.
; In such case, no map name matching is necessary and this section can be empty.
; If map names in file are different from those in DB, you should map each MAP_NAME in the file to a MAP_NAME in DB.
; The manual mapping is below:
;MAP_NAME_IN_FILE=MAP_NAME in DB
;Chr1=Chr.1
;Chr2=Chr.2
;Chr3=Chr.3
; Check 'printmapping' command that may help generating the name mapping tables.
; Alternatively, use MapIdentifiedBy
; MapsIdentifiedBy: if all maps in the file instead of the map name are identified by their alternative IDs like MAP_ID, ACCESSION_NO or GENOME_DNA_ID,
; provide the mapping with just one line using either MapName, MapId, AccessionNo or GenomeDnaId, for example:
;MapsIdentifiedBy=AccessionNo
; LoadListedMapsOnly: if true, only data for the maps listed in this section will be added.
; If false, PersephoneShell will still try to match names from the file to maps in the database
; using MAP_NAME (case-sensitive), and if the map is not found, the annotation line will be skipped
;LoadListedMapsOnly=true
[DbSequences]
; Oracle only
; The ID columns below are used in loading annotations.
; If there is no sequence/trigger assigned to these columns, you must specify a sequence for them.
;PROCESS_RUN.RUN_ID=ID_SEQ
;GDNA_ANNOT.ANNOT_ID=ID_SEQ
;DESCRIPTION.DESCR_ID=ID_SEQ
;TRACK.TRACK_ID=ID_SEQ
Please refer to the comments in the control file, they document the instruction lines in detail.
There are a few points to focus on.
- Some GFF files may have inconsistent records. For example, exons can overlap, or, a 'gene' record can cover a shorter region than the child exons, etc. Such inconsistencies are not tolerated by the loader. To allow PersephoneShell load the rest of the records, please consider using the instruction SkipInconsistentRecords:
SkipInconsistentRecords=SkipMRNA
Without this instruction, PersephoneShell will refuse loading the models if there are errors in a GFF. When reading the data from the GFF file, PersephoneShell assembles gene models from records of type 'gene', 'mRNA', 'exon', 'CDS', 'five_prime_UTR', 'three_prime_UTR'. Any inconsistency is reported. If you still want to load the file skipping the wrong records, you will need to use SkipInconsistentRecords instruction.
- It is worth noting that in case the textual value is provided as one line, the quotation marks are not necessary:
QualifierType="mRNA"
and
QualifierType=mRNA
will have the same effect. At the same time, if the text contains multiple lines, the value should be enclosed into quotation marks:
TrackDescription="Gene annotation from
some external resource.
Publication:
Authors
Title"
The text can include simple HTML tags, such as <i>, <b> or <href>. Note, that internal quotation marks needed for the HTML code should be "escaped" by backslash (\):
"Visit the main page at <a href=\"https://google.com\">Google</a>"
Qualifiers
The instruction QualifierType= specifies which lines of GFF file should be used as the source of qualifiers. If the qualifiers you plan to load appear on the lines of type 'gene' and 'mRNA', list them both as the qualifier types:
QualifierType=gene,mRNA
Gene qualifiers — key-value pairs normally stored in the GFF attribute field — can be imported using the same names as their corresponding attributes. The GFF attributes are listed in the last column of the GFF file. Here are three attributes:
ID=Glyma.01G000100.Wm82.a4.v1;Name=Glyma.01G000100;ancestorIdentifier=Glyma.01G000100.Wm82.a2.v1
To create qualifiers with the same name as the attributes, list them in an instruction that starts with QualifierAttributes=...
This will create qualifiers with the key identical to the attribute name. For example,
QualifierAttributes=ancestorIdentifier
will create a qualifier with the key ancestorIdentifier and corresponding value (Glyma.01G000100.Wm82.a2.v1). You can request more than one qualifier in this instruction:
QualifierAttributes=ancestorIdentifier,ID,Name
If QualifierType lists more than one type, there is a chance that attributes with the same name appear in both types. In such cases, to disambiguate the names, we need to use a fully-qualified form of the attribute including the type prefix, such as gene:Name or mRNA:Name.
In the test mode, you will see a line with all potential qualifiers for a given GFF type printed in the form suitable for placing into the instruction QualifierAttributes=:
Available attributes (for the INI file):
mRNA:ID,mRNA:Name,mRNA:pacid,mRNA:longest,mRNA:Parent
You can copy and paste the suggested text to the INI file:
QualifierAttributes=mRNA:ID,mRNA:Name,mRNA:pacid,mRNA:longest,mRNA:Parent
and remove the qualifiers that should be skipped.
The GFF attribute can be stored as a qualifier with a different name. In the example below, we refer to the attribute Name from the 'gene' line (gene:Name) as qualifier 'geneName'. The qualifier 'Name' from the line 'mRNA' (mRNA:Name) becomes 'transcriptName'. Once the GFF attributes have been assigned to qualifiers, reference them by the qualifier names in the further instructions, such as QualifierLinks or AnnotationSearches:
QualifierAttributeKey.gene:Name=geneName
QualifierAttributeKey.mRNA:Name=transcriptName
[AnnotationSearches]
GeneName=transcriptName
GeneFunction=Note
When running in test mode, PersephoneShell will display the list of potential qualifiers from GFF lines of the requested types specified in QualifierType= instruction. In our case, we ask to process qualifiers from types gene and mRNA:
QualifierType=gene,mRNA
The test mode will print the qualifiers found in the corresponding lines:
PS> add annotation -c Samples/Annotation/Wm82.a4_phytozome-gff.ini -t
...
List of distinct qualifiers in file (with example values):
mRNA:
ID (86,256 records): Glyma.01G000100.1.Wm82.a4.v1
[ind] [included] Name (86,256 records): Glyma.01G000100.1
[included] pacid (86,256 records): 41118016
[included] longest (86,256 records): 1
[included] ancestorIdentifier (55,193 records): Glyma.01G000100.1.Wm82.a2.v1
Parent (86,256 records): Glyma.01G000100.Wm82.a4.v1
The included qualifiers with example values will be clearly highlighted to help you see what data will be loaded and whether your selection of qualifiers needs any modification. The text [ind] marks the qualifiers that will be indexed for text search (see QualifierFilter fields in the PersephoneShell configuration file).
Note that a special qualifier is added automatically when annotations are loaded from a GFF file: parent_gene_id. It captures which gene record was the parent for mRNA records. This will be useful when exporting the gene models in GFF format and when defining the logic of gene model grouping.
Tip
If you don't yet know which attributes to load from the GFF file, specify the GFF types to collect the qualifiers from by using QualifierType but leave QualifierAttributes commented out. Then run the command 'add annotation' in test mode, which will list all the attributes collected from the qualifier types. This will help you decide which attributes to load. You can copy the text in the output that lists all qualifiers in one line and paste it to the INI file under QualifierAttributes. After that, you can remove the qualifiers that you want to skip.
GFF type
If we are talking about protein-coding genes, three types of gene model tracks can be loaded:
(a) transcripts with internal CDS;
(b) CDS only;
(c) exons only.
Normally, the gene models are loaded together with UTRs and CDS parts of the gene. In this case, keep the Type record commented out. If you prefer to load the CDS part only, so that the gene models always start with the initiator ATG codon, set Type to CDS
; Type: GFF type (column 3) of annotation items. If not specified, both exon (SO:0000147) and CDS (SO:0000316) will be parsed.
Type="CDS"
If, for some reason, you would like to load only exon structure without the CDS information, specify "exon" as the GFF type to be loaded:
; Type: GFF type (column 3) of annotation items. If not specified, both exon (SO:0000147) and CDS (SO:0000316) will be parsed.
Type="exon"
Usually, Type record is commented out, and both "CDS" and "exon" GFF types are processed.
The Type does not have to be "CDS" or "exon". For example, if you are loading miRNA track, the GFF type could be
Type="miRNA_primary_transcript"
To load multi-part records, such as pairs of primers or BLAST matches that should be chained together, explicitly specify the parent in ParentType:
ParentType=match
Type=match_part
Please also consult a sample INI file for loading micro-RNAs. Use cases are given at the bottom of this page.
Annotation groups
Gene models with CDS that share overlapping location on the genome are grouped. By default, the grouping is done based on location - the overlapping models are assigned to the same group. This behavior can be changed by forcing the process to use common qualifier values, such as values of the qualifier geneName or parent_gene_id. The qualifier parent_gene_id is automatically created during the annotation loading. It captures the information about which gene record was specified as a parent of an mRNA record in the GFF file. It is quite common to use this qualifier to form the groups: the gene models that share common values of this qualifier will belong to the same group.
GroupNameQualifier=parent_gene_id
In the downstream processing, the groups are used to store the ortholog information. Instead of recording multiple links between splice variants in a pair of genomes, PersephoneShell stores just the links between annotation groups. This significantly reduces the size of the data records in the database.
QualifierLinks
This section converts qualifiers into hyperlinks. The qualifier values, either the entire string or its part extracted by using a regular expression, will be converted into a URL.
In simple cases, use the full qualifier value to fill the placeholder %s in the URL. The instruction
Name="https://phytozome-next.jgi.doe.gov/report/transcript/Gmax_Wm82_a4_v1/%s"
will result in the value (e.g., Glyma.01G053300.1) of the qualifier Name placed entirely into the link, like this:
https://phytozome-next.jgi.doe.gov/report/transcript/Gmax_Wm82_a4_v1/Glyma.01G053300.1
Sometimes, a long text of the qualifier value contains multiple identifiers that can be used to form different URLs:
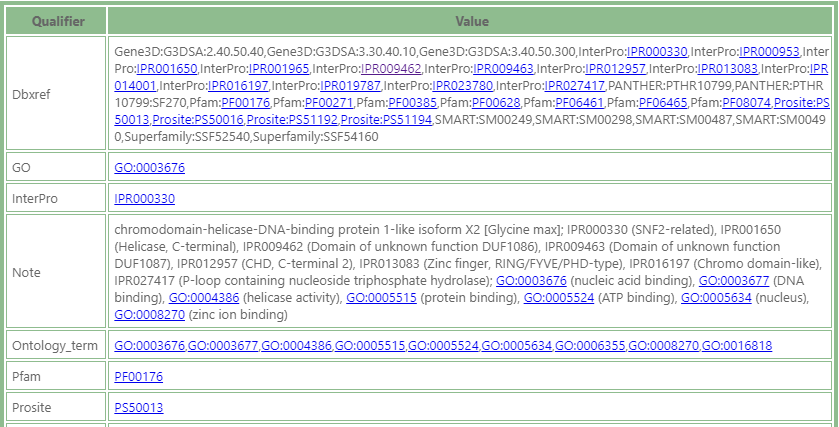
Take a look at the qualifier Ontology_term. It contains multiple identifiers, each of them worth to be presented as a separate hyperlink. To extract the proper sub-string from the text of the qualifier, use regular expressions. To generate multiple URLs from the value of Ontology_term, use this construct:
Ontology_term=http://amigo.geneontology.org/amigo/term/%s|(GO:\w+)
The regular expression (GO:\w+) specified after the pipe symbol will be used to extract the text according to the capture rule enclosed in the parentheses. Each match to GO:\w+ will form a separate link and will be embedded into the text of the value of Ontology_term.
You can apply several URL-regex pairs to the same qualifier. According to the syntax of the INI files, it is not allowed to use the same key under the same section more than once. You cannot have:
Qualifier1=link1
Qualifier1=link2
To work around this limitation, specify the qualifier name only once and provide the URL-regex pairs on separate lines:
Dbxref=http://amigo.geneontology.org/amigo/term/%s|(GO:\w+)
https://prosite.expasy.org/%s|Prosite:(PS\w+)
https://pfam.xfam.org/family/%s|(PF\d+)
https://www.ebi.ac.uk/interpro/entry/InterPro/%s/|(IPR\d+)
Just for reference, here are a few useful examples:
InterPro=https://www.ebi.ac.uk/interpro/entry/InterPro/%s/|(IPR\d+)
Pfam=https://pfam.xfam.org/family/%s|(PF\d+)
Prosite=https://prosite.expasy.org/%s|Prosite:(PS\w+)
GO=http://amigo.geneontology.org/amigo/term/%s|(GO:\w+)
Dbxref=http://amigo.geneontology.org/amigo/term/%s|(GO:\w+)
https://prosite.expasy.org/%s|Prosite:(PS\w+)
https://pfam.xfam.org/family/%s|(PF\d+)
https://www.ebi.ac.uk/interpro/entry/InterPro/%s/|(IPR\d+)
Remember that you can always add more qualifier links after the gene annotation has been loaded. Use the command 'add qualifier_link' or 'edit qualifier_link'
MapMapping
Let's focus on MapMapping section. This division is sometimes necessary if the GFF file references chromosomes using names that are different from their names in the database. For example, the sequences in the database can have names such as 'Chr.1', 'Chr.2', etc., while the records in the GFF file use '1', '2', etc. Or you can imagine when the original FASTA file has complex definition lines that use quite-uninformative accession names together with human-readable strings like 'chromosome 1'. During loading, the sequences are given names like 'chromosome 1' but the GFF file may reference them by the accession number. The MapMapping section allows to associate the sequence names in the file to the map names in the database. The table may list the names in the file and provide the corresponding name in the database. The maps in the database can be identified by MapName, AccessionNo, GenomeDnaId, or MapId. You can generate such translation table by PersephoneShell's command printmapping. If you know that '1' in the file corresponds to the map called 'Chr.1' in the database, put a record
"1"="Chr.1"
Note, the double quotes are not necessary if the names do not contain spaces.
Please see other methods of the map mapping in the Control files section.
Under Annotation, find Sources line and use the correct path to the GFF file.
As usual, first, test the INI file by using '-t' switch:
PS> add annotation -c add_MSU_osa1r7.ini -t
and, if all tests are successful, load the data, usually with the verbose output:
PS> add annotation -c add_MSU_osa1r7.ini -v
Use cases
Loading regular gene models
If the GFF file contains gene models only, you can leave ParentType and Type commented out. The hierarchy of parent-child relationships for the gene elements will be detected automatically. The gene models are assembled from the records of well-established sequence ontology types 'gene', 'mRNA', 'exon', 'CDS', 'five_prime_UTR', 'three_prime_UTR'. Sometimes, the child records are placed under the parent record of type 'transcript'. In most cases, you do not need to provide any additional instructions: ParentType different from 'mRNA' will still be detected if all gene parts use the standard records, such as 'exon' and 'CDS'.
Loading matches
The GFF3 files sometimes include BLAST matches for protein homologs or primers. The matches may contain multiple parts. Specify the entire structure that you want to capture in the instructions ParentType and Type, for example:
ParentType=primer_match
Type=match_part, match
Loading problematic GFF files
Sometimes, the GFF files contain systematic errors. For example, there can be duplicated or misplaced exons. You still can load such gene models if you explicitly instruct the program to assemble the gene models from types that have no errors. If 'exon' records should be ignored, list the other gene types in the Type instruction:
Type=CDS,three_prime_UTR,five_prime_UTR
Note that the rest of the models in the GFF file still can be loaded with IgnoreInconsistentRecords set to SkipMRNA, which will force the program to skip erroneous gene models.
 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.