Add Map Set, Map, and Sequences
Once the Organism record is created, you can start adding map sets. To add these components to an organism data set, you need to use the add command (see Add) pointing to a control file (see Control Files), which specifies how the data files should be processed.
In Persephone data model, a map set contains maps. Maps contain tracks. The map sets can be of two main kinds: genetic (linkage groups) and physical (based on a sequence). To create map sets and maps for genomes that have genomic sequences, use the add sequence command. The control INI file for this kind of load will have all the necessary information to create map set with empty maps (no tracks yet). Each map set should have Display Name (MapSetName - unique label under parent node in the map set tree), MapSetAccession (unique across the entire database), OrganismId (existing in the database), the place in the map set tree and the source (the institution name that published the maps).
The map set is placed in the tree shown in Persephone main application on the left. The tree resembles folder structure, which suggests the way to form the full path of a map set in the tree. For example, "/Arabidopsis thaliana/TAIR10" is the path to the map set TAIR10 placed under parent node "/Arabidopsis thaliana".

This section shows an example with the steps necessary to add a map set, maps, and sequences for version a4 (Wm82.a4.v1) of Glycine max genome. For the full description of various parameters of the add sequence command please consult this page.
Before adding genomic sequences in FASTA format, we need to list the FASTA headers to decide which method should be used to extract the map names. Quite often the FASTA headers are short and descriptive enough to be used directly as the map names:
$ less Gmax_508_v4.0.softmasked.fa.gz |grep ">"
>Gm01
>Gm01_scaffold_166
>Gm02
>Gm02_scaffold_122
>Gm03
>Gm03_scaffold_135
>Gm03_scaffold_238
>Gm03_scaffold_245
>Gm03_scaffold_27
>Gm03_scaffold_521
>Gm03_scaffold_926
>Gm04
>Gm05
...
Note
We recommend using the command 'analyze fasta <path-to-fasta-file>' to list the headers and length of each sequence:
PS> analyze fasta /data/Data/soy/Gmax_508_v4.0.softmasked.fa.gz
LENGTH HEADER
-----------------------------------------------------------------------------------------------------------------------------------------------------------
57,932,355 Gm01
45,523 Gm01_scaffold_166
50,400,358 Gm02
60,768 Gm02_scaffold_122
46,951,866 Gm03
54,556 Gm03_scaffold_135
Our strategy in this case is to copy the entire FASTA header into the map name. In more complex cases, we apply regular expressions or split the FASTA header to extract the map names. But for his particular example with the soybean sequences, we will take the FASTA headers as a whole and save them as the map names. We do not have to split anything or use regular expressions.
We will use a similar approach to get the AccessionNo: without additional instructions the entire FASTA header will be saved as the accession number.
As for the chromosome name, we will use a regular expression to extract the numeric part of the map name, such as 01 in Gm01. The term ChromosomeName is a short name used for the maps that represent genome - many of them are shown together in the graphical view of the map set, so the names should be short:
Open a copy of the Wm82.a4-phytozome.ini control file, which is included in the PersephoneShell "Samples/Sequence" folder and is shown below.
[ProcessRun]
; RunDescription: if specified, a custom description will be used,
; otherwise, "Added sequences for {MapSet Accession No.} from {Sources}." will be used.
RunDescription="Loaded sequences for Wm82.a4.v1 from Phytozome"
[MapSet]
;------------------------------------------------------------------------------------------------
; 1. Using existing MapSet
;------------------------------------------------------------------------------------------------
; If either MapSetId or MapSetPath is specified, it adds sequences to the existing MapSet.
; otherwise, a new MapSet should be specified below.
; MapSetId: id of an existing map set
;MapSetId=270000441
; MapSetPath: path of an existing map set.
;MapSetPath="Glycine max/Wm82.a2.v1"
;------------------------------------------------------------------------------------------------
; 2. Adding new MapSet
;------------------------------------------------------------------------------------------------
; Organism ID (required): organism ID should exist.
OrganismId=3847
; Display name (required): a name shown in MapSetTree. Usually a assembly build name.
DisplayName="Wm82.a4.v1"
; Description: by default, organism name + display name. Note that the internal quotation marks should be escaped using backslash: \"
Description="<i>Glycine max</i> Wm82.a4.v1 from Phytozome.
Downloaded from https://data.jgi.doe.gov/refine-download/phytozome?organism=Gmax&expanded=508
Project description: <a href=\"https://phytozome-next.jgi.doe.gov/info/Gmax_Wm82_a4_v1\">https://phytozome-next.jgi.doe.gov/info/Gmax_Wm82_a4_v1</a>
"
; AccessionNo (required): accession of the genome build. See http://ncbi.nlm.nih.gov/genome
AccessionNo="Wm82.a4.v1"
; Source ID: database or institution that the MapSet/sequence originate. One word, no spaces.
SourceId="Phytozome"
; MapSetQuality: value between 0 and 4 describing the completeness and quality of the genome assembly
; Depending on this value, different signal-bar icons will be used in the map set tree
MapSetQuality=4
[MapSetTree]
;------------------------------------------------------------------------------------------------
; 1. Adding new MapSetTree node to a parent node
;------------------------------------------------------------------------------------------------
; Parent node ID: if specified, the MapSet with the new sequences will be placed under this parent node as a child.
;ParentNodeId=200206082
;------------------------------------------------------------------------------------------------
; 2. Adding new MapSetTree node under a new root node
;------------------------------------------------------------------------------------------------
; Root node name: usually an organism name. Ignored if the root name already exists. A longer path consisting of more than one node can be specified here. The nodes will be created starting with the root node.
;RootNode="Plants/Glycine max"
RootNodeName="Glycine max"
; Root node order number: order of the root node in the MapSetTree. By default, 0.
RootNodeOrderNo=0
; OrderNo: placement of map set under its parent node in the map set tree. If not specified, OrderNo will be automatically incremented for each new map set.
; If you want to order all map sets under a parent node alphabetically, set OrderNo to a fixed value. All map sets will get the same value and will be sorted alphabetically by name.
;OrderNo=0
[Sequence]
; Sources (required): FASTA file(s) of genomic DNA sequence located locally or remotely accessible via URL.
Sources=$DATA/soy/Gmax_508_v4.0.softmasked.fa.gz
; Commit frequency: commits after reading this many nucleotides. Large numbers requre larger rollback segment, smaller numbers result in higher frequency of transactions
CommitFrequency=10000000
; ExcludedHeadersRegEx: exclude sequences that match this regular expression
;ExcludedHeadersRegEx="chloroplast"
; IncludedHeadersRegEx: include only sequences that match this regular expression. Note, ExcludedHeadersRegEx, if specified, is applied first
;IncludedHeadersRegEx="complete"
; FASTA header starts with '>' and provides map information delimited by delimiters follows (Delimiter=VerticalBar):
; >{0}|{1}|{2}|{3}|{4}
; Delimiter: specify one among Colon(:), Comma(,), Period(.), Hyphen(-), SemiColon(;), Slash(/), Tab(\t), Space( ) and VerticalBar(|)
; Comment this line out if the FASTA header will not be partitioned
;Delimiter=VerticalBar
; Map name index. Tells which field between the delimiters corresponds to the map name. 0-based. If delimiter is not used, MapNameIndex is ignored,
; the entire FASTA header is saved as the map name.
;MapNameIndex=0
; If the part between delimiters is too long to be stored as the map name, it can be parsed and the map name can be extracted by using this regular expression
; If MapNameIndex is not provided, MapNameRegEx is applied to the whole FASTA header
;MapNameRegEx=".*chromosome (..).*"
; ReplaceSpacesWith: Provide symbols that will be used to replace spaces in the map name. Some third-party tools are confused if the map name contains a space.
;ReplaceSpacesWith=_
; Map accession index. Tells which part between delimiters corresponds to map accession. 0-based. If delimiter is not used, MapAccessionIndex is ignored,
; the entire FASTA header is saved as the map accession.
;MapAccessionIndex=0
; If the part between delimiters is too long to be stored as the map accession, it can be parsed and the map accession can be extracted by using this regular expression
; If MapAccessionIndex is not provided, MapAccessionRegEx is applied to the whole FASTA header
;MapAccessionRegEx=".*chromosome (..).*"
; Map description index
;MapDescriptionIndex=0
; MapDescriptionRegEx: regular expression to extract the map description from FASTA header or, if MapDescriptionIndex is provided,
; from the extracted field at that index
;MapDescriptionRegEx="(.*)"
; MapDescriptionExtra: extra text appended to the description that can potentially embed map name ($MAPNAME) or accession ($MAPACCESSION) to form a hyperlink.
;MapDescriptionExtra=<a href=\"https://www.ncbi.nlm.nih.gov/nuccore/$MAPNAME\" target=\"_blank\">NCBI</a>
; Some sequences can be called chromosomes, some as scaffolds. Usually, the number of chromosomes is small and allows to show all of them as a representation of a genome
; ChromosomeCriteriaRegEx - if the fasta header matches the RegEx criteria, the entry will be called a chromosome
;ChromosomeCriteriaRegEx="chromosome"
; ChromosomeCriteriaLength - a formula to separate chromosomes from the rest of the sequences. It can be an alternative
; to ChoromosomeCriteriaRegEx
; For example, ChromosomeCriteriaLength=">5000000" will store sequences longer than 5,000,000 bp as chromosomes
ChromosomeCriteriaLength=">5000000"
; In case the sequence passes the criteria to be called a chromosome, the chromosome name will be read from the field with this index:
;ChromosomeNameIndex=0
; ChromosomeNameRegEx - a regular expression applied to the field with the given index to extract the chromosome name
ChromosomeNameRegEx="Gm(.+)"
; Expected length index: if specified, the expected length of each sequence will be compared to the actual sequence length.
;ExpectedLengthIndex=5
; MapNameFilterRegEx: Regular expression filter based on map name: include only sequences whose map name matches the given pattern.
; if not specified, all the sequences in the source will be included.
; Example below would load only sequences with name that start with 'Chr.'
;MapNameFilterRegEx="^Chr\..*"
; Length filter: used to include only sequences whose length satisfies the criteria. Use '>' or '<' to load sequences longer/shorter than the given number (e.g,"<1000000")
; if not specified, all the sequences in the source will be included.
;LengthFilter=">1000000"
; ChromosomeReorder: force ordering chromosomes. By default (None), chromosomes are shown in the order they appear in the file.
; If ChromosomeReorder is Natural, chromosomes are ordered in natural order (chr1, chr2, ..., chr10, chr11, ...).
; If ChromosomeReorder is ByLength, chromosomes are ordered by length, longest to shortest.
;ChromosomeReorder=Natural
[TranslationCode]
; List map names and their corresponding translation codes. Default code is 1
;Pt=11
; Specify parameters to calculate the GC histograms
[Gc]
; what percentage must 'N' take for a region to be considered poly-N, in range[1-99]. Default is 25
;PolyNPercentage=50
; Histogram step (size of a 'cell'), in range [1-127], the lesser the value the more data will take space but the more precise it will be. Default is 100
;HistogramStep=100
Important
The SourceId field (set to "Phytozome" in the example above) cannot contain spaces.
Note that the source file can be fetched via a URL from a remote location. If you have a slow network connection or work with large files, it is recommended to download the file first and use its local copy, as this file is accessed at least twice: during the test phase (using the flag -t) and when uploading it to the database (-v). Sometimes, as in the case of Phytozome, pre-downloading files is the only way, since transferring the file must be done using the Phytozome's interactive pages.
When referencing a local file, it is highly recommended to use the PersephoneShell's variable $DATA pointing to the data root folder. The variable is set in the configuration file. The path to the referenced local file would look like:
Sources=$DATA/soy/Gmax_508_v4.0.softmasked.fa.gz
PersephoneShell can recognize archive files compressed with gzip. Note, that rarely, the tar.gz files with deeply nested folder structure are not recognized properly. In such case, please un-compress the file using gzip (and tar) program and reference the uncompressed file. The Sources record can also list multiple files, separated by comma or, instead of listing separate files, you can specify a folder. All files in that folder will be processed sequentially. It is also possible to load files from remote locations via http or ftp. In this case, please make sure that the URL starts with the protocol name, like 'http://'.
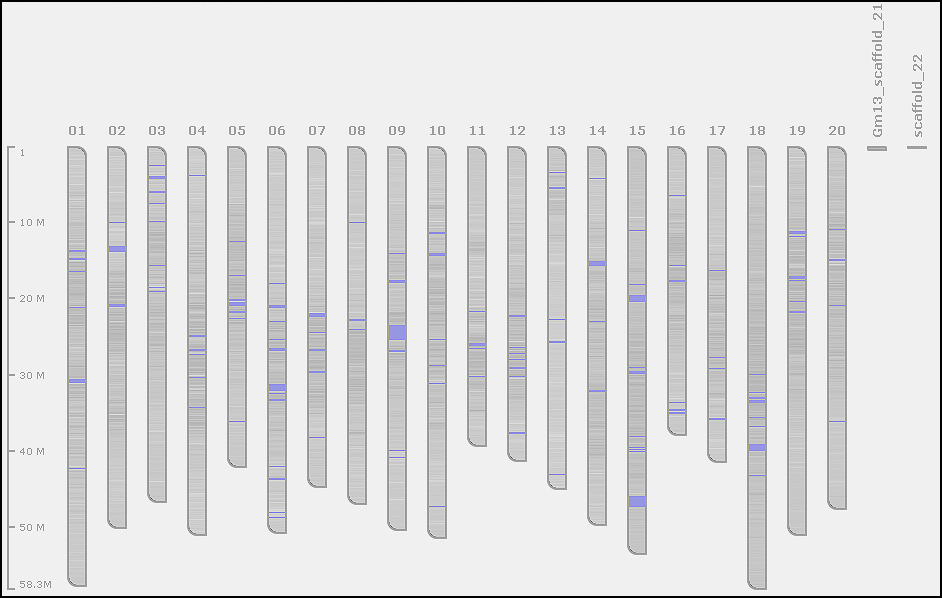
In our exercise, the FASTA file has 282 sequences: 20 regular chromosomes and additional short scaffolds. We want to mark sequences from Gm01 to Gm20 as chromosomes. This would help Persephone to correctly present the soybean genome consisting of 20 chromosomes. You will see the graphical representation of the genome once the map set is selected in the tree, so chromosome names will be shown simultaneously for multiple maps, and we recommend to make them as short as possible.
Some map sets can contain a mixture of a few chromosomes and thousands of scaffolds, so knowing which sequences should represent the core genome is important to avoid cluttering the graphics - scaffolds will be hidden by default. When listing the maps of a map set, Persephone will first show the maps marked as chromosomes preserving the order of sequences in the file, the rest of the maps (most probably scaffolds) will be sorted by length (starting with the longest ones).
By default, if we do not provide any parsing instructions, the map names and accession numbers will be copied from the entire FASTA headers. To designate chromosomes, we need to use some criteria distinguishing chromosomes from non-chromosomes. The chromosomes can be marked based on the sequence length or by finding a specific pattern in their FASTA header using regular expressions (the full logic is presented here).
In the current example, we will use ChromosomeCriteriaLength record to specify the rule to differentiate chromosomes from the rest of sequences. The rule
ChromosomeCriteriaLength=">5000000"
nominates maps longer than 5,000,000 bp as chromosomes.
Please also see how we extract the chromosome name (not to be confused with the map name) using a regular expression:
ChromosomeNameRegEx="Gm(.+)"
As we are trying to make the chromosome names as short as possible, we are removing the common prefix 'Gm' and use the chromosome name in a form '01', '02', etc.
Controlling the data flow
Just for reference here, when loading the genomic sequences, you can control:
- what sequences should be loaded (filter by name or by size): IncludeHeadersRegEx, ExcludeHeadersRegEx, LengthFilter.
- what part of the FASTA header will become the map name: MapNameIndex, MapNameRegEx.
- what part of the FASTA header will become the map accession number: MapAccessionIndex, MapAccessionRegEx.
- what part of the FASTA header will become the chromosome name: ChromosomeNameIndex, ChromosomeNameRegEx.
- what sequences will be marked as chromosomes: ChromosomeCriteriaLength, ChromosomeNameRegEx.
(Please consult another example of parsing more complicated FASTA headers).
 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.