Add Orthologs and Paralogs
Once the gene models have been loaded, they can be used to calculate orthologous gene pairs. The pairs of homologous genes can be also found between the genes in the same genome (paralogs).
The orthology can be found by running the command 'create ortholog'. PersephoneShell will prepare the protein sequences from the loaded gene models and run DIAMOND or BLASTP to find matches between two sets of genes for the two selected genomes. The simplest command will calculate paralogs by referencing a single map set:
PS> create ortholog <mapSetId>
For example, if the MapSetId of our genome is 111, the command will be:
PS> create ortholog 111
This will find the best reciprocal matches for every gene among other genes of the same genome. For each gene, the program will try to find the best matching protein. Apparently, the best match to a given protein is the query protein itself. When searching for paralogs, PersephoneShell excludes self-matches and matches to its splice variants.
Once the paralogs have been calculated, they can be revealed in the Synteny Matrix. Each dot represents an ortholog gene pair plotted at the corresponding coordinates for two genes:
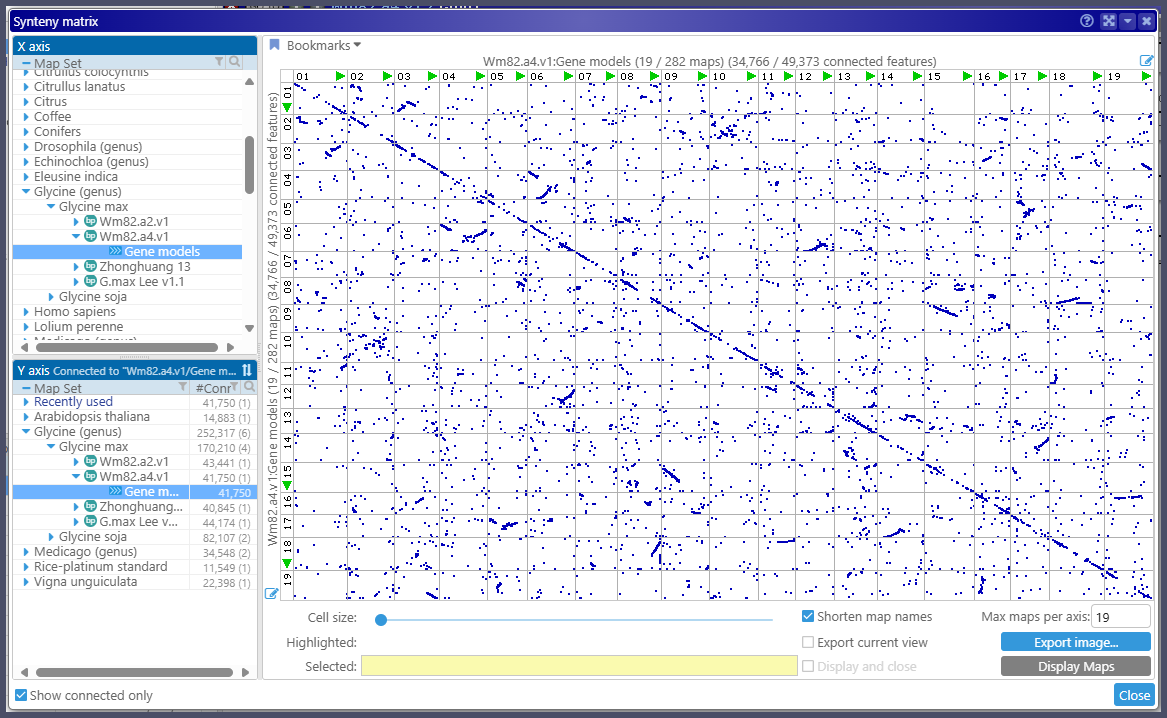
Note that, despite the fact that self matches are excluded, the paralogs appear on the main diagonal, which nicely reflects the tandem gene duplication.
To calculate the orthologs between different map sets, type the additional MapSetId(s) on the command line. To identify orthologs between map set 111 and 222 run:
PS> create ortholog 111 222
If you want to find orthologs to multiple genomes, this can be done in one command. The parameter for MapSetId(s) accepts ranges of IDs, for instance:
PS> create ortholog 111 10..15,24,26
will find orthologs between map set 111 and map sets 10,11,12,13,14,15,24 and 26.
 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.