Adding Orthologs
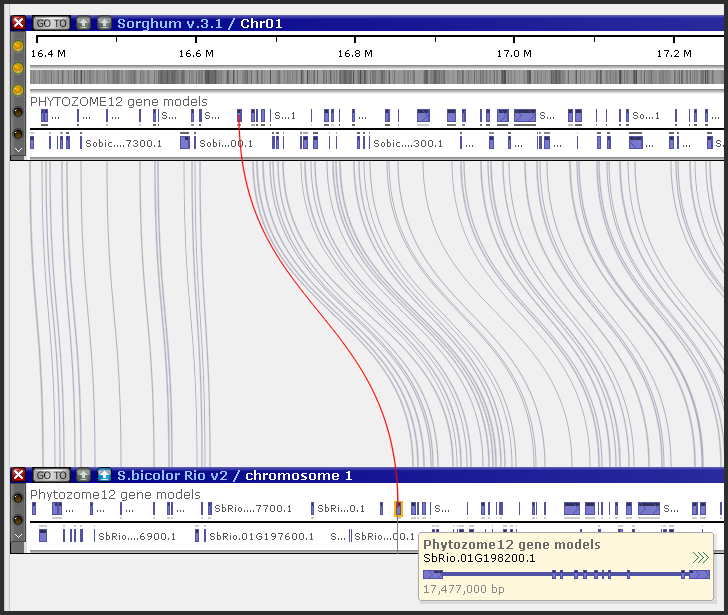
Persephone connects orthologous genes by a thin connector line.
The links between genes, stored in the database, can be derived by using different methods outside of Persephone. The database would just store the information that a pair of proteins are somehow related, most commonly because they are highly similar. If a protein B is the best match for a given protein A and, reciprocally, protein A is the best match for protein B, such genes will be considered orthologs. The database can also store other matches with lower similarity score, but only the best matching proteins will be called orthologs and shown linked by a connector line. Ortholog pairs are also used to generate the Synteny matrix.
In the gene properties form, the orthologs are shown in a separate tab:
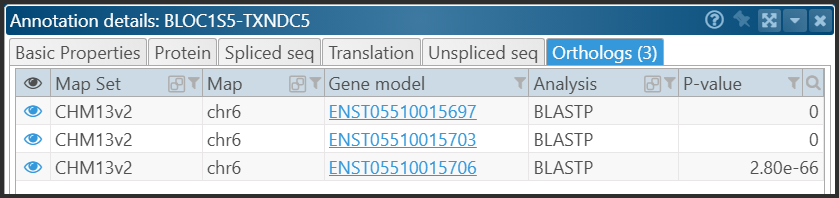
Orthologs can be calculated by PersephoneShell (create ortholog) or, if the pairs of genes have already been derived and stored in a file, they can be added by using the command
add orthologs -c ...
Data files
To load the orthologs, prepare a file with several columns separated by TextDelimiter specified in the INI file. PersephoneShell recognizes several formats:
- AnnotId - 5 columns (Tab-separated by default): AnnotId1, AnnotId2, Score, Evalue, PercentIdentity
The first two columns are required, the other 3 are optional. AnnotIds are unique identifiers assigned by PersephoneShell to the protein's original gene model during loading the annotation. The FASTA file with proteins for BLAST use AnnotId in the header for each sequence. This allows using the existing BLAST index files for calculating the orthologs identified by AnnotId.
;sample
123456 98765 123 0.0 99.4
234566 98766 90.5 1e-6 96.2
...
- MapSetAccGeneName - 4 columns (Tab-separated by default): MapSetAccession1, GeneName1, MapSetAccession2, GeneName2
All 4 columns are required. A pair of MapSetAccession and GeneName should uniquely identify a gene model. GeneName is a value stored as one of the gene's qualifiers. The name of such qualifier should be either specified (GeneNameQualifier=) in the INI file for loading orthologs or, if it is not provided, a record in ORGANISM_CONFIG table will be used to find the qualifier that stores the gene name (see Adding annotation, section [AnnotationSearches]).
;sample
TAIR10 AT1G07110.1 MSU_osa1r7 13105.m00781
TAIR10 AT1G07120.1 MSU_osa1r7 13103.m02191
...
- MapSetNameGeneName - 4 columns (Tab-separated by default): MapSetName1, GeneName1, MapSetName2, GeneName2
All 4 columns are required. A pair of MapSetName and GeneName should uniquely identify a gene model. The logic is similar to the item above, just instead of map set accession, the map set name is used. - OrthologUpdater (Comma-separated by default) - the format produced by the 'create ortholog' command and consumed by a separate tool called OrthologUpdater, that compresses the orthologs into binary blocks for faster retrieval. The format requires 4 header rows defining the map sets by their AccessionNo (which is guaranteed to be unique) and track names for the two gene sets being compared:
#fromMapSet:TAIR10-1
#fromTrack:Ensembl
#toMapSet:ASM465v1
#toTrack:BGI gene models
AT1G01020.1,BGIOSGA009702-TA
AT1G01030.1,BGIOSGA011738-TA
AT1G01040.1,BGIOSGA011556-TA
AT1G01050.1,BGIOSGA019932-TA
Please note that the sharp (#) symbol is a part of the instruction, not a comment.
As an option, you can add pvalue as the last column to the file in OrthologUpdater format:
#fromMapSet:TAIR10-1
#fromTrack:Ensembl
#toMapSet:ASM465v1
#toTrack:BGI gene models
AT1G01020.1,BGIOSGA009702-TA,0.001
AT1G01030.1,BGIOSGA011738-TA,1e-100
Control files
The INI control file is quite simple (please read the comments):
[ProcessRun]
RunDescription="Load orthologs between rice japonica and rice indica"
[Orthologs]
; Delimited text file; one line per ortholog link
Source="$DATA/orhologs/orthologs_jap_ind.txt"
; Method used to derive the ortholog pairs
Analysis="BLASTP"
; Input format (required) can be of one of: AnnotId, MapSetAccGeneName, MapSetNameGeneName or OrthologUpdater
; AnnotId - first two columns are required but 3 more are optional. AnnotIds represent protein's original gene model
; AnnotId1, AnnotId2, Score, Evalue, PercentIdentity
; MapSetAccGeneName - 4 columns are required:
; MapSetAccession1, GeneName1, MapSetAccession2, GeneName2
; MapSetNameGeneName - 4 columns are required:
; MapSetName1, GeneName1, MapSetName2, GeneName2
; OrthologUpdater (comma-separated by default) - special format accepted by OrthologUpdater tool for binary compression. It can also be produced by 'create ortholog' command
; see https://help.persephonesoft.com/AddingOrthologs.html for the description of format
InputFormat=MapSetAccGeneName
; TextDelimiter: specify one among Colon(:), Comma(,), Period(.), Hyphen(-), SemiColon(;), Slash(/), Tab(\t), Space( ) and VerticalBar(|)
; The default TextDelimiter for most of the formats is Tab.
; OrthologUpdater format uses Comma by default
;TextDelimiter=Tab
; If GeneNameQualifier is not specified (see below), make sure that ORGANISM_CONFIG (see annotation_search) contains an entry that specifies which qualifier stores gene name,
; which would allow to unambiguously convert gene name to ANNOT_ID
; GeneNameQualifier - use this qualifier to uniquely find gene by its name (applied for both map sets). If the qualifier is not provided, ORGANISM_CONFIG entries will be used
GeneNameQualifier=locus_tag
; Instead of GeneNameQualifier, which specifies common qualifier storing gene names, GeneNameQualifier1 and GeneNameQualifier2 can be used
; for the genes in the left or in the right columns respectively.
;GeneNameQualifier1=transcriptName
;GeneNameQualifier2=Name
; TrackName1 and TrackName2 can be used in addition to GeneNameQualifier if the gene name belongs to genes in more than one track.
; For example, you can have two tracks "CDS" and "MRNA" that show the genes with the same name nomenclature. So, there could be more than one
; gene model corresponding to a given name. Use TrackName to disambiguate
;TrackName=CDS
All formats except 'AnnotId' reference the genes by their name. The gene name is stored in one of annotation qualifiers. If this qualifier is specified in annotation_search for gene names, PersephoneShell will try to match the names to their unique AnnotIds automatically. However, if this information is not provided in the database, you will need to specify this qualifier name explicitly using the GeneNameQualifier instruction. If the two map sets use different gene name qualifiers, they should be provided as GeneNameQualifier1 and GeneNameQualifier2. Such qualifier will be looked up in all gene models for the given map set, which can include several tracks. If there is an ambiguity, and one gene name corresponds to two or more gene models from different tracks, another instruction TrackName (or TrackName1, TrackName2) limiting the search to a given track will be required.
 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.