Create Function
The command create is used to generate new data based on the records in the database. One of the data types that can be created is functional annotation. We can use well characterized protein sequences, such as SwissProt, to assign the functional annotation to the proteins that show a good match. For each gene in a track we can find the best matching protein from SwissProt with defined function somewhat implying that the gene performs a similar function.
The command
create function <mapset> [-a]
will find matches between the proteins from an annotation track and SwissProt. (We decided to leave 'function' instead of 'functional_annotation' just to save some typing).
Note
Before running the command create function please make sure that you have installed diamond (install diamond) and SwissProt (install swissprot)
If there are more than one annotation track with CDS, the program will ask to choose a track for the analysis:
PS> create function "Rice-platinum standard/Os GJ-temp: IRGSP-1.0 (Nipponbare)"
Assigning functional annotation by match to well-characterized proteins from SwissProt
Testing ...
- SwissProt file is present: passed
- DIAMOND binaries found: passed
- Working on map set Rice-platinum standard/Os GJ-temp: IRGSP-1.0 (Nipponbare)
More than one annotation track is found for map set Rice-platinum standard/Os GJ-temp: IRGSP-1.0 (Nipponbare). Which track should be used?
[0]: MSU gene models
[1]: Gnomon gene models
[2]: FGENESH gene models
[3]: Gramene Gene models
[4]: CSHL Gene models
Enter [TrackNumber] to use?
A flag -a can be used on the command line to force processing of all annotation tracks without asking.
As usual, the map set can be provided via MapSetId or its MapSetPath, such as in the example above. The auto-complete feature will save quite some typing.
The test mode (-t) will check for the presence of the binaries of diamond and the SwissProt index. The queries for diamond are the protein sequences for the annotation track in question. They are extracted from the corresponding BLAST index files and saved in a form of a FASTA file. When the test reports success, run the analysis, usually in verbose mode (-v).
Running diamond for one track usually takes about half a minute. The matches (one best match per gene) will be stored as an annotation qualifier 'SwissProt match':
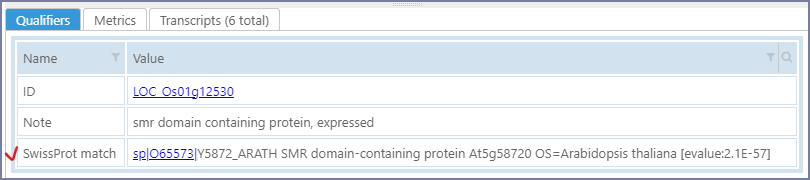
At the end of the processing, PersephoneShell will nominate this qualifier as containing the Function of a gene. This distinction is useful when searching for gene properties - the users can narrow down the search to searching in Function qualifiers only, thus reducing the chance for false positive hits. Internally, PersephoneShell will add a new annotation_search entry. Please see the section for adding annotation_search for more details.
 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.