Create Synteny
This command will run minimap2 to find synteny between all maps of two genomes.
create synteny <mapSetId1> <mapSetId2> [-p <minimap_parameters>]
The default parameters for running minimap2 (-x asm5 -t 4 -m 1000 -N 2 -k 28) used by PersephoneShell set the limit on the minimal ribbon score (-m 1000). This threshold filters out a large number of secondary connectors and tries to leave only wide ribbons that show a "big picture":
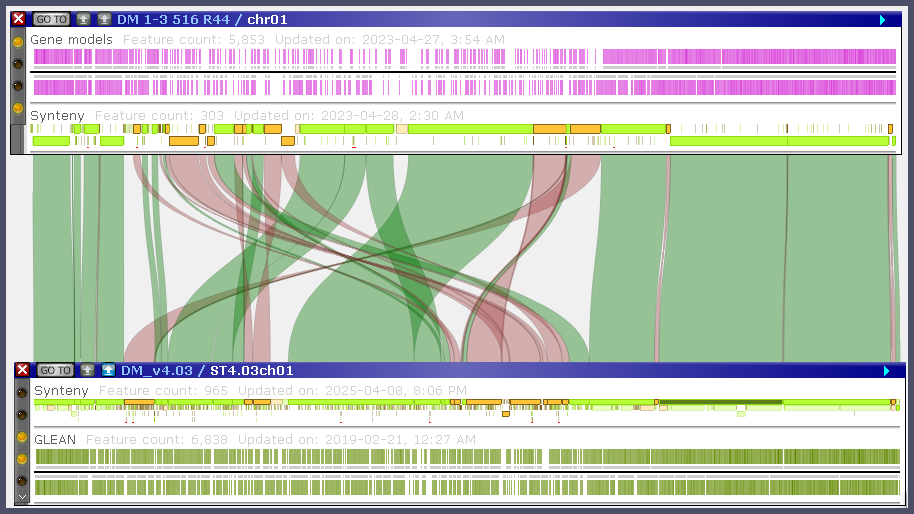
Please also note that the search is filtering out secondary matches for the query segments (-N 2). This means that if a segment on the query map shows some match, other secondary matches of this segment will not be reported. The parameter -k 28 enforces search to start with a high-identity seed where all 28 bases match perfectly. This reduces the sensitivity and greatly improves the search performance. You can change this value by providing your number after -k, but this also means that the other parameters from the default preset should be explicitly repeated in the command's parameter -p (see below).
The results of minimap2 are saved in a temporary *.paf file and stored in the database. The synteny ribbons are "anchored" to the track called "Synteny".
The default parameters can be changed by using the command line parameter -p (or --params):
create synteny 11 22 -p "-x asm5 -t 8 -m 10000"
Please watch the number of produced ribbons as it will affect the graphical performance of Persephone. We don't expect any issue if the synteny contains a few thousand of ribbons.
If minimap2 exits with an error, most likely it ran out of memory (Exit code:137). Try to reduce the number of threads (the parameter -t, which is set to 4 by default). You can limit the amount of memory used by minimap2 by adding the parameter -K. To set the limit to 500 MB, use -K 500M.
The asm5 preset in minimap2 is designed for long assembly-to-reference mapping with sequence divergence below 5%. Here are the parameters equivalent to asm5:(note that parameter pairs can be used without space: -k19 is the same as -k 19)
-k19: K-mer size of 19
-w19: Minimizer window size of 19
-A1: Match score of 1
-B19: Mismatch penalty of 19
-O39,81: Gap open penalties of 39 (short gap) and 81 (long gap)
-E3,1: Gap extension penalties of 3 (short gap) and 1 (long gap)
-s200: Minimum chaining score of 200
-z200: Z-drop score of 200
-N50: Maximum number of chains per query of 50
--min-occ-floor=100: Minimum occurrence threshold of 100
These settings ensure that alignments do not extend into regions with 5% or higher sequence divergence. If your sequences have higher divergence, you might want to consider asm10 or asm20 presets instead.
Please consult the documentation for minimap2.
 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.