Mapping Primers for PCR Products
Using BLAST to search for primer hits can be tricky, especially when you expect mismatches. This section provides guidelines and examples to improve your results.
First, please keep in mind that the default NCBI blastn evalue parameter is 10. (Evalue is a number of "random" hits (occurrences of the query sequence) that one should expect for a BLAST data file of given size that occur purely by chance.) The smaller the size of genome, the smaller is the chance to find the query sequence. When searching for short queries, make sure that evalue is large enough; otherwise, the hits may be filtered out. Persephone uses evalue of 1000 in its "Primer" mode.
Second, to initiate a match, BLAST needs "seeds" of N consecutive identical nucleotides. The BLAST parameter word_size controls the value of N. You can see this in the parameters text box of Persephone's BLAST interface. If your primer is 20 nucleotides long and you are searching for a perfect match, you will need to make sure that the word_size parameter is set to 20 or lower.
Moreover, if you want to allow one mismatch (95% identity for 20 bp primer), you cannot just set the perc_identity parameter to 95 because you still need to watch the seed size. With the seed of size 20 and a primer with one expected mismatch, there will be no seeds (BLAST would require a stretch of 20 identical nucleotides first), so the filter of 95% would be applied to an empty list.
For example, the mismatch occurs in the middle of the primer. This means that to initiate the match you would require a seed of not more than 10 nucleotides.
CGATCTTGCGAGGCTTACGC
||||||||| ||||||||||
CGATCTTGCTAGGCTTACGC
The seed is highlighted above. Once the seed is found, BLAST would try to extend it on both ends to find the boundaries of the match. Thus the whole primer would be matched with one mismatch.
To remove multiple false positives, we recommend using two primers as shown below:
>primer1
Atgtcggcggcggcgaaggc
>primer2
tcagtcatcatcttgaccag
Use them both as a query as shown below.
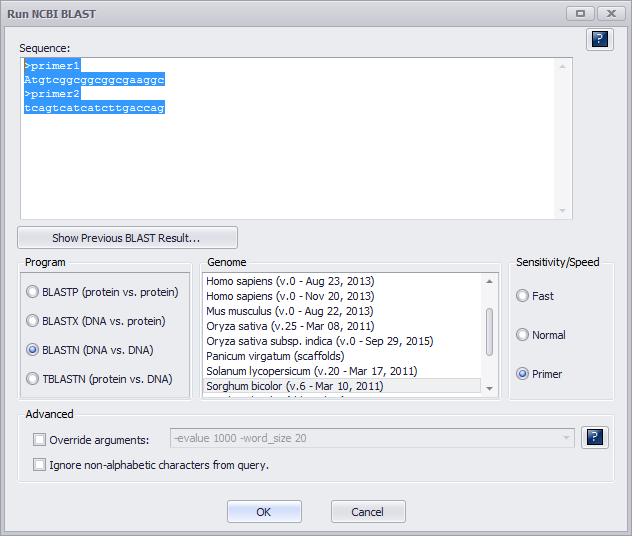
Note
You need to ensure the Primer radio button under Sensitivity/Speed has been selected as shown above.
In case of "perfect" primers, using default Primer parameters (-evalue 1000 –word_size 20) should work as shown below.
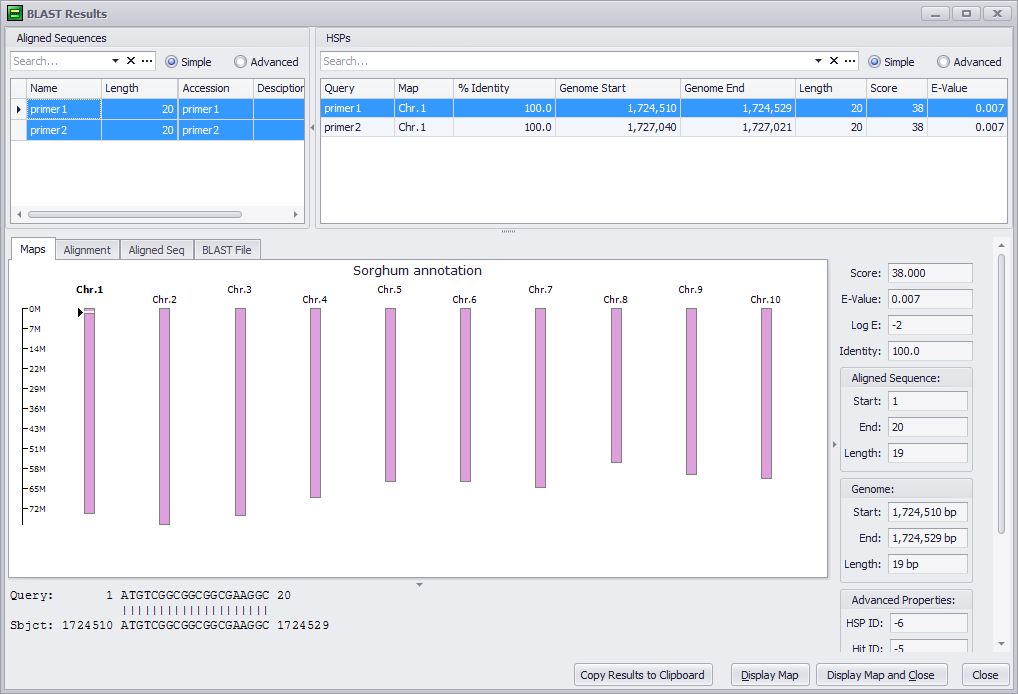
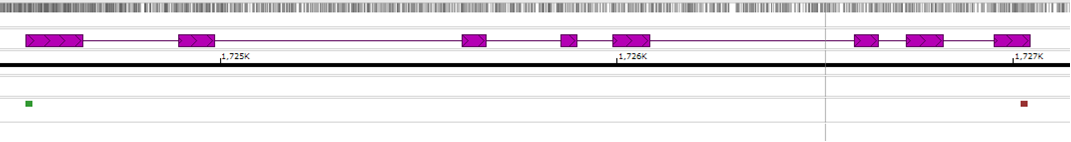
Confirm the primer mapping by clicking the Display Map and Close button and then switching to horizontal view. You should see both primers in the newly-created BLAST track (green:forward, red:opposite):
If one mismatch is expected, then word_size should be reduced to 10. Select (check) Override arguments under Advanced as shown below.
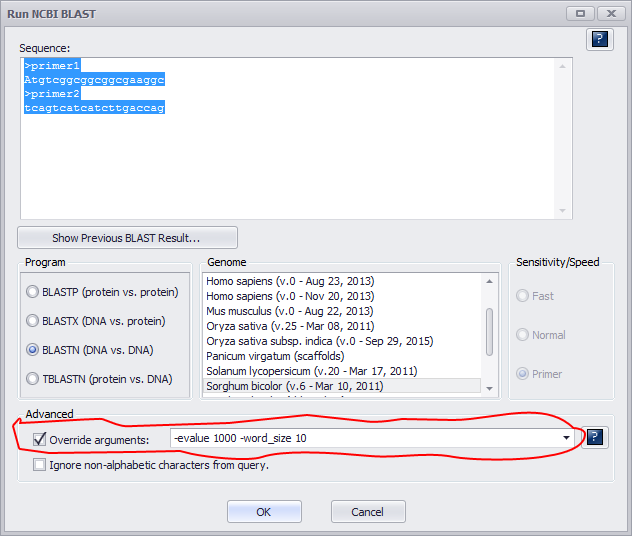
Reducing the size of seed would generate a lot of hits. In this example we are looking for a pair of matches (primer1 and primer2) located closely on the same chromosome.
To find good pairs you could use advanced filter. Click the Advanced radio-button in the grid as shown below.

Next, click Edit Filter to open the Filter Editor as shown below.

Specify values for %Identity filter and Length of the match. Next, sort by Map and Genome Start to see if any hits are located close to each other on the same chromosome. Be sure to select both primer1 and primer2 queries in the top left panel.
Tip
See Using Advanced Filters for more information about the Filter Editor.
 Copyright © 2009-2024 by Persephone Software. All Rights Reserved.
Copyright © 2009-2024 by Persephone Software. All Rights Reserved.