Web Persephone: The BAM Read details dialog
This dialog displays detailed information about a single BAM (CRAM) read, as shown on the BAM track. To open the dialog, click on any read on the BAM track. While the dialog is open, the read will be outlined with a thick border:

The BAM Read Details dialog is split into two halves: the top pane, and the bottom pane; you can drag the splitter bar with the mouse to resize them. Both panes contain several tabs. By default, the Basic Properties tab will be selected on the top, and the Auxiliary tags tab will be selected on the bottom; however, the dialog will "remember" which tabs were selected the last time it was opened, and select the same tabs the next time it is opened.
Top: Basic Properties
This tab displays all the basic properties of the selected BAM read.

|
Read Name: |
The name of the currently selected read (e.g. as provided by the sequencer). |
Mapping quality: |
The quality of the read, as recorded in the BAM file. |
|
Location: |
The start and end coordinates of the read's alignment on the map (in bp). Click the |
Mate read: |
Displays the location of the current read's paired mate (this box will be empty if the read is unpaired). Click the location text to open the BAM Read Details dialog for the mate; click the If the paired mate is located on another map, its location will be highlighted in orange: |
|
CIGAR string: |
The CIGAR string encodes the gapped alignment of this read (as described in e.g. the SAMTools specification). If the string is too long, you can click the |
Read length: |
The length of this read, in nucleotides. |
|
SAM flags: |
Binary flags describing some additional properties of the read; these are shown in detail on the Binary flags tab at the bottom. |
Organism: |
The organism to which BAM file belongs. |
|
Map Set: |
The map set where this BAM file belongs. Click the map set name to open its Map Set details dialog; click the |
Map: |
The map where this BAM file belongs. Click the map name to open the Map details dialog; click the |







Top: Sequence
This tab displays the full sequence of the BAM read.
The full CIGAR string is listed at the top, followed by the color-coded FASTA record of this read's sequence. You can use the column width controls in the bottom-left corner of the pane to wrap the sequence to a specific character length (in the same way as it is done in e.g. the Annotation Details dialog). The display options on the right-hand side control the formatting and highlighting of the sequence.
- Strand, Direct/Reverse: Reverse-complements the sequence; the original strand of this read's alignment is indicated in parentheses.
- Highlight: Toggles highlighting options for this read.
- Base modification (5mC): This option is only available if 5mC (methylation) data is present in the BAM file (inside the MM/ML fields). When this option is selected, nucleotides in the sequence are highlighted just as they are on the BAM track: blue if the probability of methylation is high, and red if it is low.

- CIGAR feature: Highlights nucleotides according to their status in the CIGAR string; options that are not present in the current BAM read are disabled.
- Mismatch: Single nucleotides that do not match the reference sequence are highlighted according to their base.
- Insertion: Highlights insertions in the BAM read relative to the reference sequence.
- Deletion: Highlights deletions; if the Show ref. base option is checked, bases in the reference sequence are substituted in place of the usual dashes:


- Padding: Highlights long padding sequences (e.g. in case of intron-spanning reads), similarly to how small deletions are highlighted:
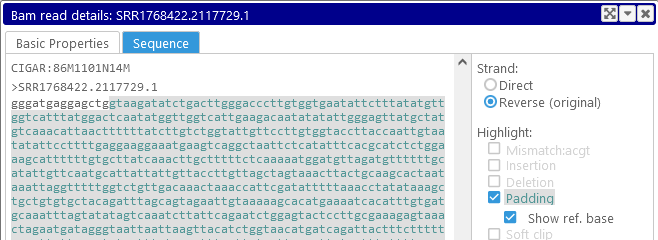
- Soft clip: Highlights soft-clipped fragments of the read:

- Quality threshold: If read quality information is present in the BAM file, you can use this slider to adjust the quality threshold. Bases whose quality is above the threshold will be capitalized; lower-quality bases will remain in lowercase. Currently, this feature is disabled for reads longer than 1,000 nucleotides (for performance reasons). Note that you can also view detailed quality information on the Base qualities tab in the bottom pane.
You can hover the mouse cursor over any base in the sequence to view a tooltip listing additional information:


The tooltip displays the value of the base in the BAM read (if any), the value of the corresponding reference base (if any), and the exact position of the base in the read as well as on the reference map; and finally the CIGAR operator that pertains to the highlighted position on the BAM read.
Bottom: Auxiliary tags
This tab lists all the auxiliary tags for this read, as provided in the BAM file.

As usual, you can sort and filter these tags using Persephone's standard table sorting and filtering controls. By default, values that are too large to fit in this view are abbreviated, but you can expand them by clicking the  button; click the
button; click the  button to collapse the text once more:
button to collapse the text once more:

You can also click the  button to copy the entire tag value to the clipboard (regardless of whether it is expanded).
button to copy the entire tag value to the clipboard (regardless of whether it is expanded).
Bottom: Binary flags
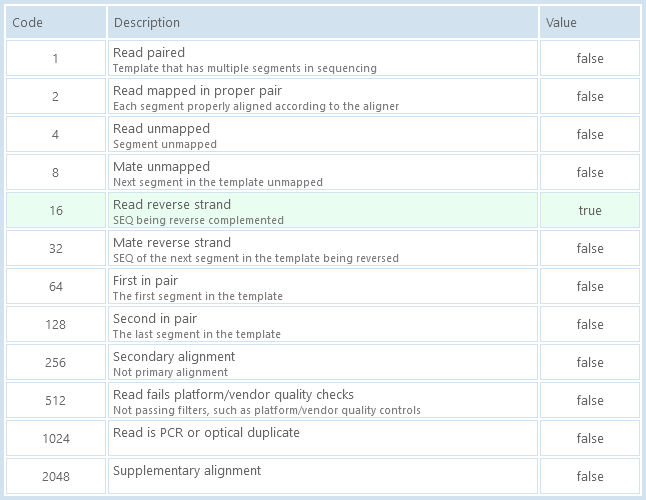
This tab lists all the binary flags for the current BAM read. Flags that are set to true are highlighted in light green:
Bottom: Base qualities
This tab is available only if base quality information is present in the BAM file. It displays the histogram of base quality values across the entire read sequence, as well as the ASCII-encoded quality string:

Select the Raw data option to instead view decoded base quality values as a comma-separated list:

 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.