Web Persephone: The BAM track
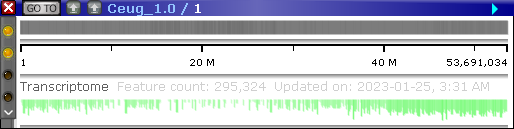
The BAM track displays multiple reads aligned to a reference genome. In collapsed mode, the track only shows the distribution of reads as a histogram:
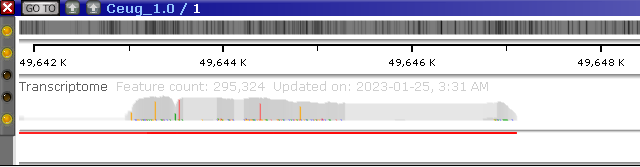
Expand the track and zoom in to show the pile-up of reads in more detail. The track will automatically adjust its height (similarly to the Annotation track):
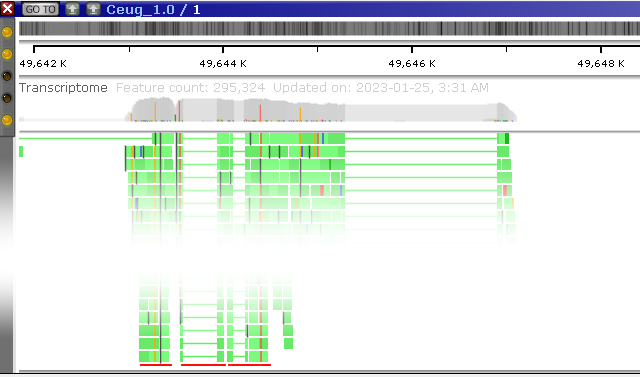
Note
This high-resolution preview is not available for BAM files that were imported into into the current browser session instead of long-term private storage.
The red lines at the bottom of the track indicate that some of the reads extend beyond its edge. You can manually adjust the track's height by dragging its bottom edge with the mouse:
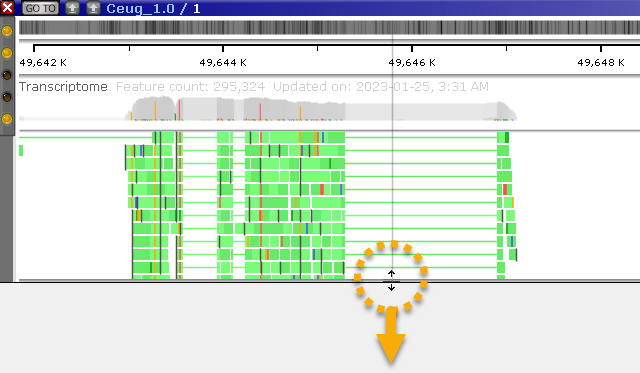
As you drag, the track will adjust the thickness of the reads to fit the available space as tightly as possible.
You can also manually increase or reduce the thickness of the reads by choosing the appropriate option from the track's right-click context menu, or by holding down the Alt key and rolling the mouse wheel. For example, you could make the reads thinner to view more of them at a glance in the pile-up:
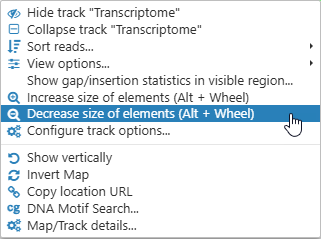
Note
The Alt + Mouse wheel shortcut may not work properly in Firefox.
Zoom in to view individual read alignments in more detail:

The coverage histogram
The coverage histogram at the top of the track displays summary information about the reads in the pile-up. You can drag the thick line below the histogram to make it wider or narrower:
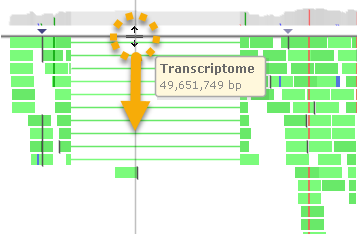
The dark gray areas on the histogram indicate aligned reads; light gray areas indicate gaps or deletions. Colored lines indicate single-nucleotide mismatches, which are colored according to the base pair on the read: a c g t .
By default, the histogram is displayed on the logarithmic scale. To change the scaling, right-click anywhere on the track, then choose View options, and then set the desired scale: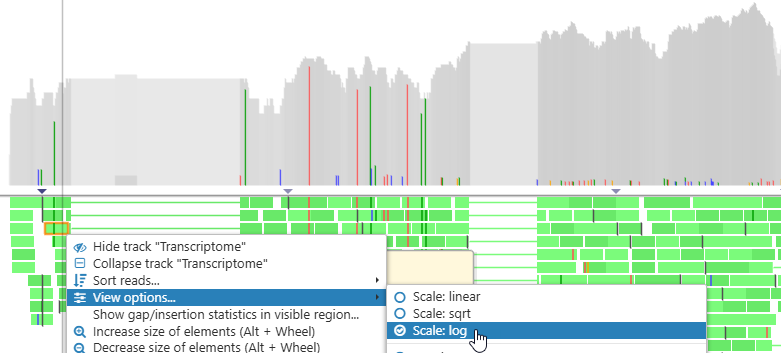

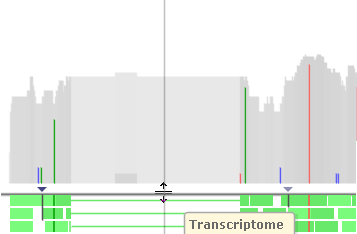
The blank line underneath the histogram is reserved for insertion indicators: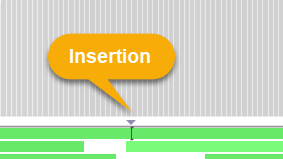
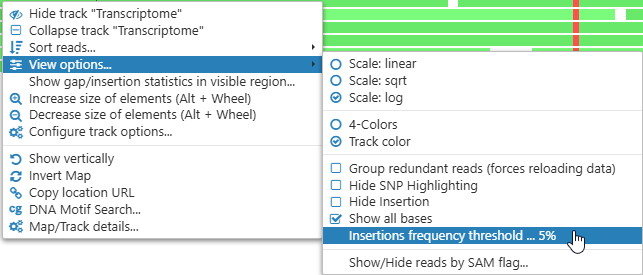
These downward-facing triangles indicate that there's an insertion somewhere in the read pile-up at the given position (the insertions themselves are rendered as I-shaped lines on top of the reads). To reduce clutter, insertion indicators are only shown when at least 10% of the reads at the given position contain insertions. You can change this threshold in the View options menu:
Mouse over the histogram to view a tooltip balloon listing summary statistics for all the reads at the location under the mouse cursor, including the relative frequency of mismatches, insertions, deletions, and gaps: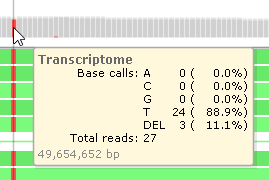
The read pile-up
BAM Reads are displayed in a pile-up underneath the histogram: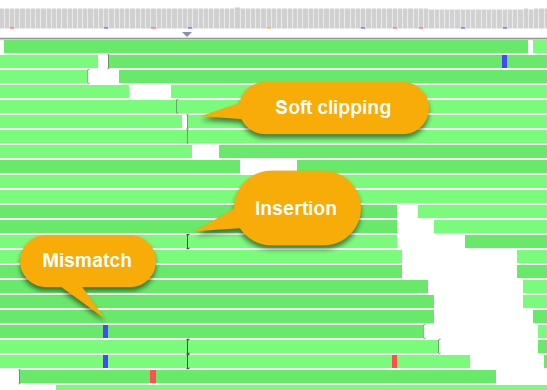
Reads on the direct strand (+) are shown in a lighter color, while reads on the reverse strand (-) are darker; gaps in read alignment are rendered as thin lines (similarly to introns on the Annotation track). By default, mismatches are highlighted according to the nucleotide that differs from the reference, although you can change this in the View options menu (discussed in more detail below).
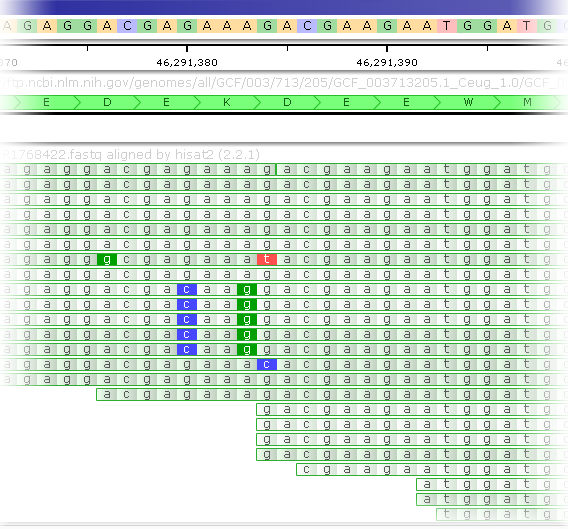
Zoom in further (and increase the thickness of track elements, if needed) to see individual nucleotides for each read; you may find it useful to also enable the GC-content track and any Annotation tracks on the same map:
Insertions are indicated by I-shaped lines on top of the reads (placed between the affected nucleotides). You can mouse over the insertion symbol to view the inserted sequence:
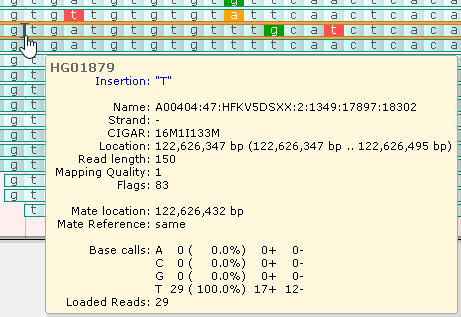
This tooltip balloon appears whenever you mouse over a read, which is then outlined with a yellow border. The balloon shows the name of the read; its CIGAR string; and additional information about its mapped location on the reference. The table of base calls (at the bottom of the balloon) lists the counts and percentages of reads with mismatches at the current location (i.e., the nucleotide currently under the mouse cursor):
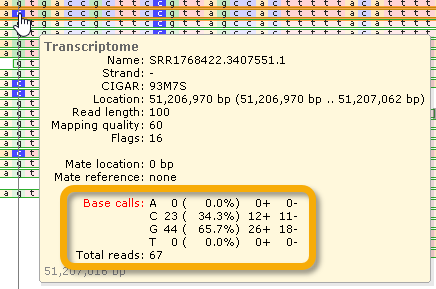
In this example, 23/67 reads have a "C" at the current location, and 44/67 reads have an "A". The table header is highlighted in red since not all reads agree on the nucleotide (indicating a potential variant call). The two rightmost columns display the counts of reads per strand.
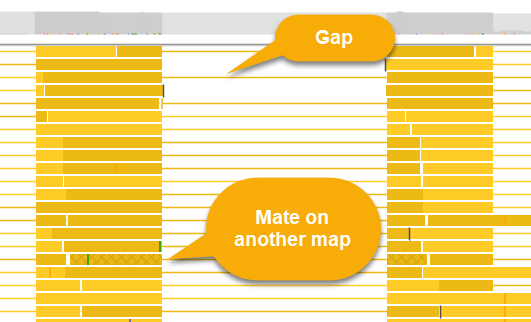
If the current read has a mate pair, its location will likewise be shown in the popup balloon; if the paired read is visible on the screen, it will be outlined with a dark red border:
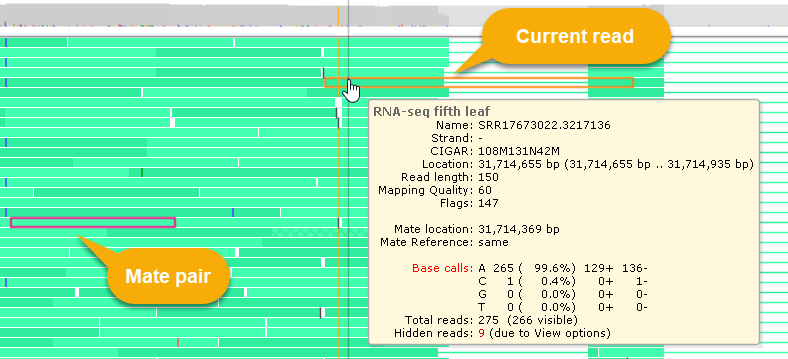
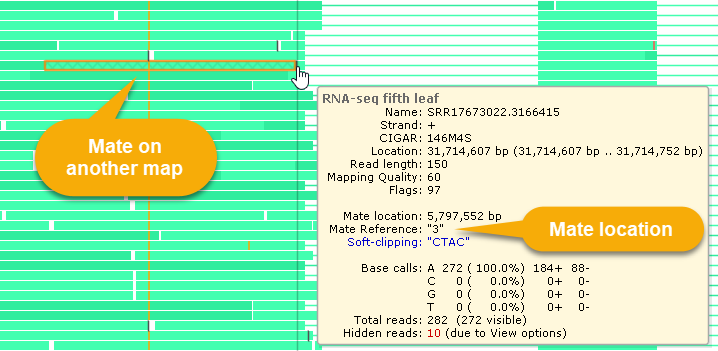
Reads whose pairs are located on another chromosome are displayed with a cross-hatched pattern, as shown above.
Deletions and padding (e.g. for intron-spanning reads) are indicated by dashes:



Soft-clipping sites are marked with square brackets; mouse over them to view the soft-clipped sequence fragment of the read:
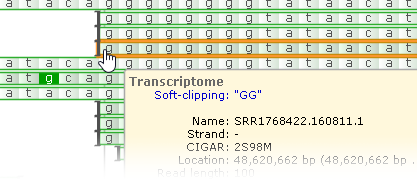
You can also click the read to open the BAM Read Details dialog, where you can view detailed information about the read and its sequence.
Methylation color coding
Some BAM files contain additional data (in MM/ML fields) indicating the level of DNA methylation (5mC) of CpG islands. By default, the nucleotides of such BAM reads are color-coded by methylation probability (blue if the probability is low, and red if it is high):
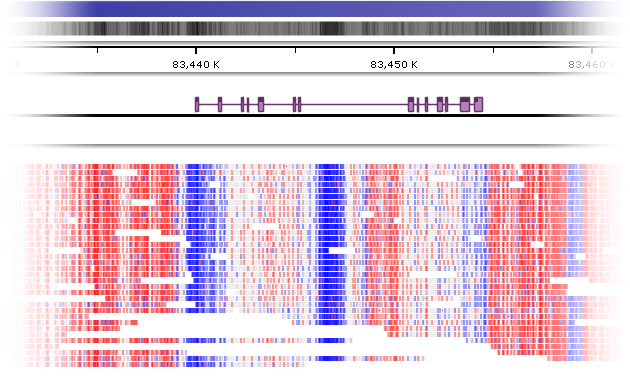
You can turn this color coding on or off in track view options, as described below. You can also preview the MM/ML fields in the read's tooltip, or click the read to view them in more detail.
Sorting reads
By default, reads in the pile-up are sorted by their coordinate (as they are in the original BAM file). To change the sort order, right-click a location on the track, and select Sort reads from the context menu. For example, you can sort the reads by base call at the chosen location: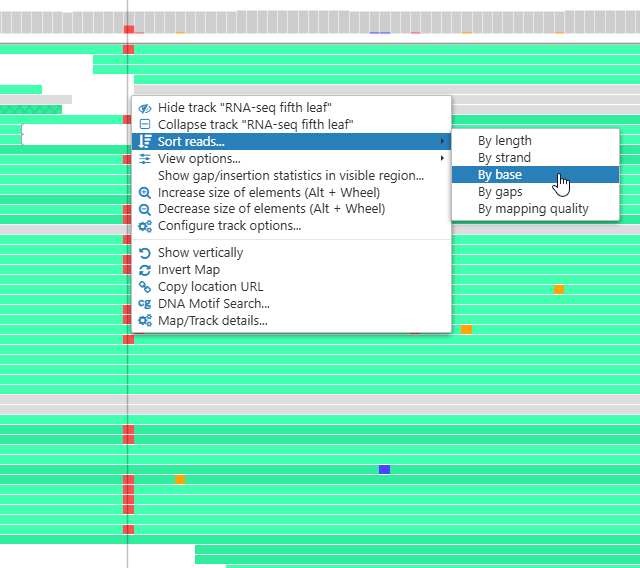
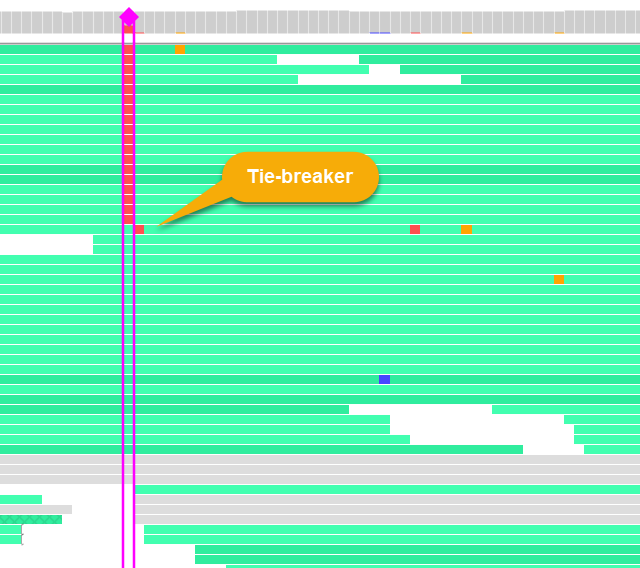
The location chosen for the sort is bracketed by pink vertical lines, and indicated by a pink diamond on the histogram. Access the Sort reads menu again to sort by a different metric, or to change the sort order (ascending vs. descending). In case of ties, neighboring locations will be used as tie-breakers (as in the example above).
View options
To change the way aligned reads are displayed, zoom in until individual reads are shown, then right-click anywhere on the BAM track and select View options from the context menu:
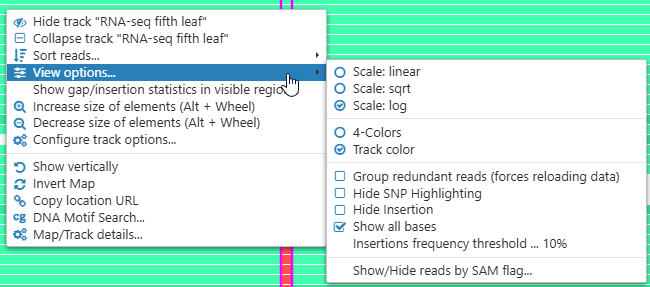
- Scaling: linear / sqrt / log. Changes the scaling mode of the histogram (as well as the preview when the track is collapsed or zoomed out). The sqrt (square root) mode offers a good balance between legibility of small values and overall appearance of peaks and valleys.
- Coloring: 4-Colors / Track color / Base Modification (5mC): Toggles coloring options of individual nucleotides on aligned reads. By default, the bases that match the reference sequence are colored according to the foreground color of the BAM track; choose 4-Colors to instead color each nucleotide according to its value. If methylation data is present in the BAM file, you can also select Base Modification (5mC) to color the nucleotides by their methylation probability:
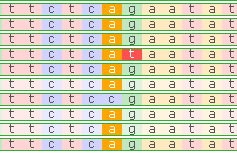

Nucleotides that are different from reference are highlighted in a bolder color; however, you can change this using the Hide SNP Highlighting option. - Group redundant reads: Collapses all reads whose locations and CIGAR strings are identical into a single element on the screen. This mode is especially useful for viewing large BAM files with many reads in the pileup:

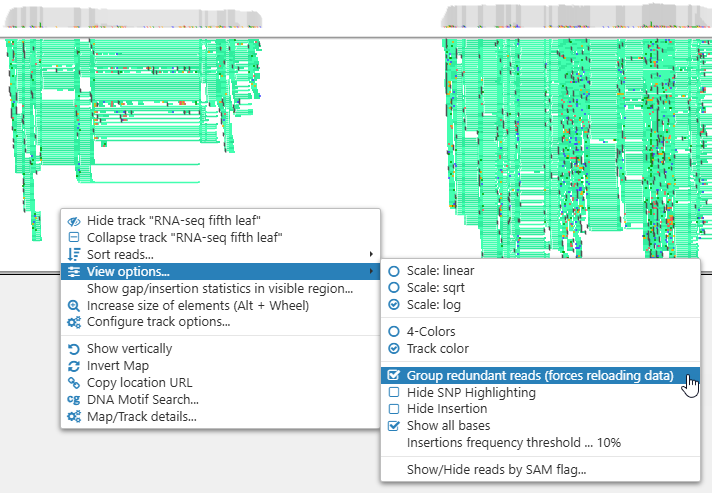
In this mode, a read's tooltip balloon will display the number of redundant reads that have been grouped together with it: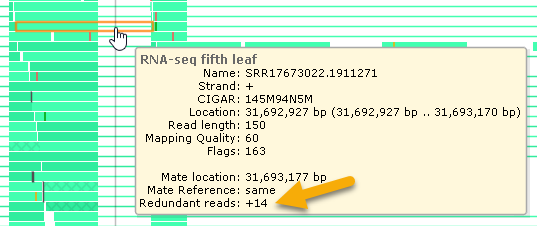
- Hide SNP Highlighting: Removes highlighting for nucleotides that are different from reference.
- Hide Insertion: Hides the insertion symbols. When using the Base Modification mode, the insertion symbols are automatically hidden as typically, PacBio long reads used to produce the modbam files have high level of errors, and the large number of insertions would obscure the picture.
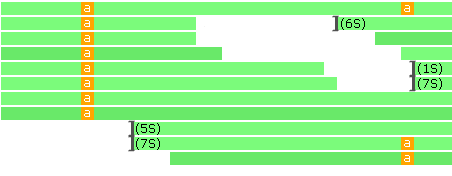
- Show all bases: Hides nucleotide letters that are identical to reference even when the track is zoomed in; doing so can make variants easier to notice. In addition, soft clipping sites are annotated with the count of soft-clipped nucleotides (in CIGAR format):
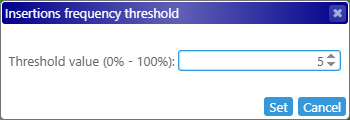
- Insertions frequency threshold: Hides insertion indicators on the histogram for those locations where the relative frequency of insertions falls below this threshold.
- Show/Hide reads by SAM flag: Opens a popup window where you can toggle read visibility according to their SAM flags, as described below.
Read visibility
Select Show/Hide reads by SAM flag from the View options menu to display a popup window listing all of the binary flags for each read (as specified in the BAM format specification):
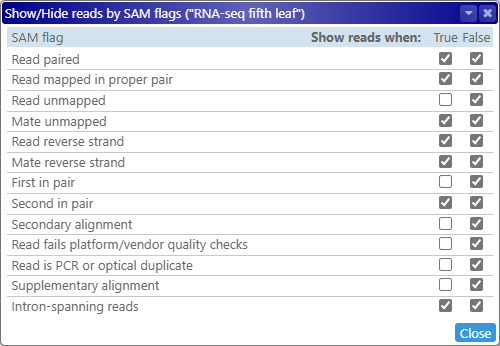
You may find it useful to dock this window to the side of the screen, so that it does not overlap the BAM track. You can toggle the True and False checkboxes to show or hide the reads matching the specified arrangement of flags. For example, by toggling the Read reverse strand flag, you could show only the reads that are aligned to the reverse strand:
...or only those reads that are aligned to the forward strand:
...or all reads regardless of strand (as is the default):
To view detailed information about a read's binary flags, click it to open the Read details dialog, then switch to the Binary flags tab. Persephone provides additional functionality for some of the flags:
- Read unmapped: Identifies the reads whose location on the map is known, but the CIGAR string is not. Such reads are hidden by default, but if you choose to display them, they will be shown in gray:
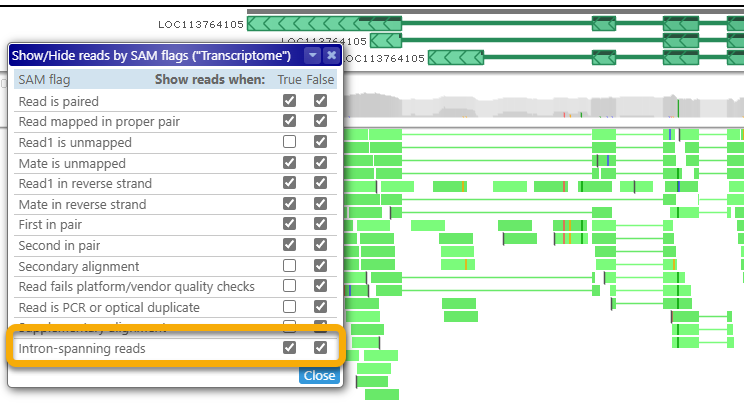
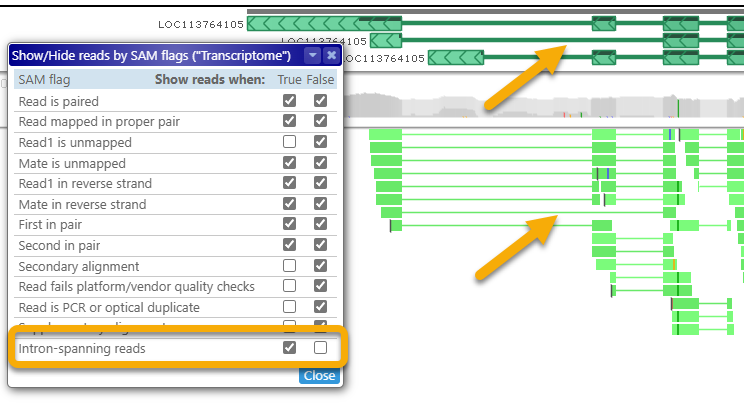
- Intron-spanning reads: Shows or hides reads with long gaps in their alignment. For example, if you are trying to validate alternative isoforms of a gene, you could show only the intron-spanning reads, and hide the rest to reduce clutter:
Gap/insertion statistics
To view detailed statistics regarding insertions and deletions on reads in a particular region of the map, first zoom in until no more than 2Mbp are visible on screen, then right-click and choose Show gap/insertion statistics in visible region from the popup menu:
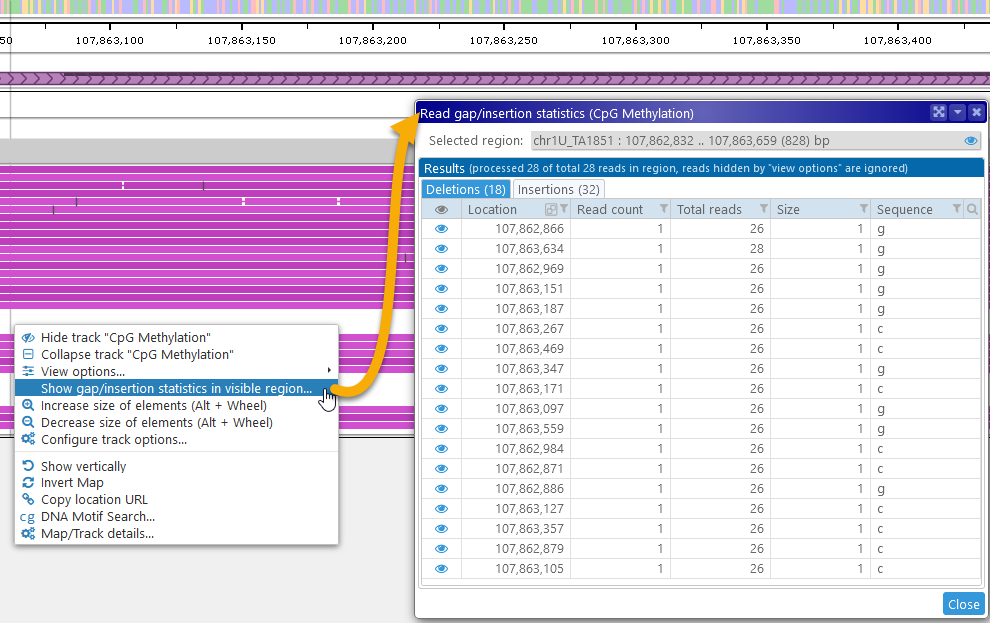
The Gap/insertion statistics dialog displays a table of all the deletions in its first tab, and a similar table on the second tab. The tab headers display the total number of deletions/insertions (in this case, 18 and 32 respectively). As usual, this table can be sorted and filtered; for example, you could show only the deletions of "g":
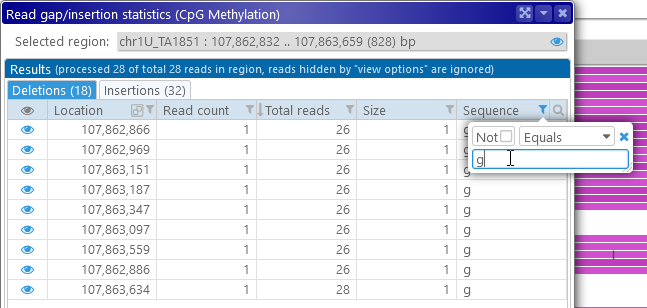
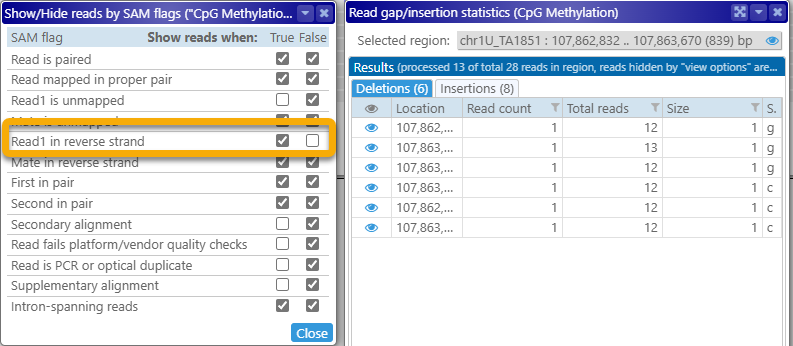
As mentioned at the top of the dialog itself, any reads that are hidden by the currently selected view options (as described above) will not appear in these tables; for example, you could hide all the reads aligned to the reverse strand: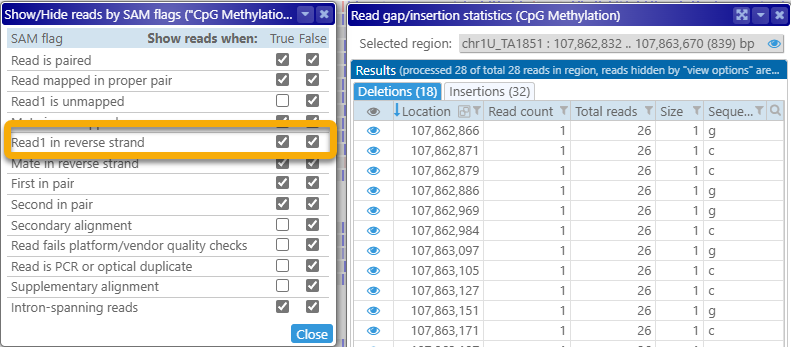
Click the  icon next to each deletion or insertion to zoom the map to that location, revealing individual nucleotides on the GC-Content track:
icon next to each deletion or insertion to zoom the map to that location, revealing individual nucleotides on the GC-Content track:
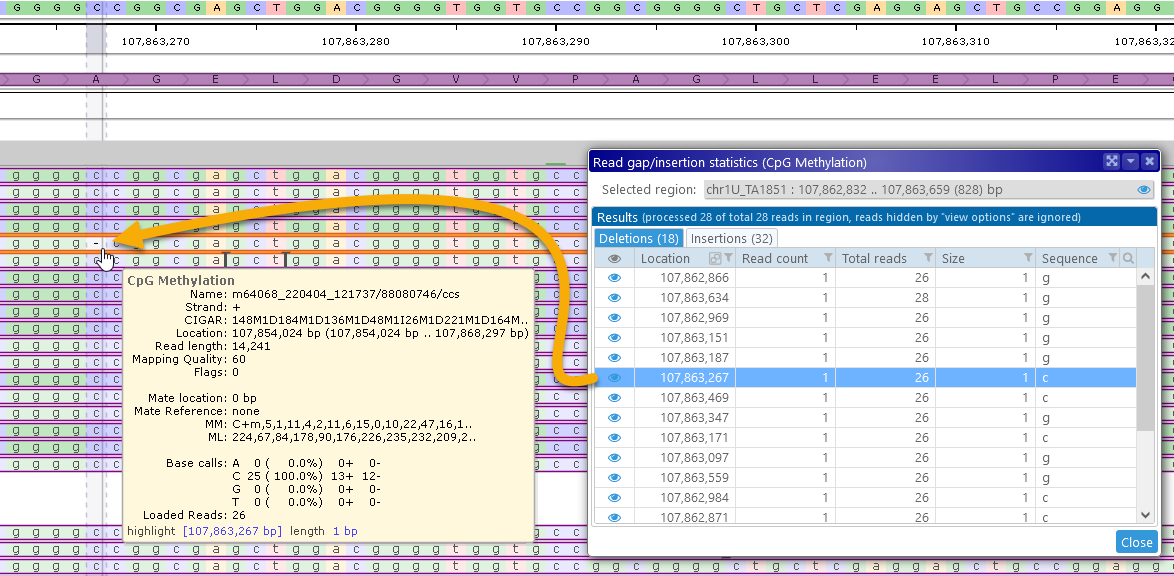
The clicked location will also be marked by a temporary highlight. To return to the original search region, click the next to the Selected region box (at the top-right corner of the dialog).
Note that this feature also works on imported BAM files; for example, here's a visualization of mutations induced by CRISPR on human stem cells, as shown on a drag-and-dropped BAM file: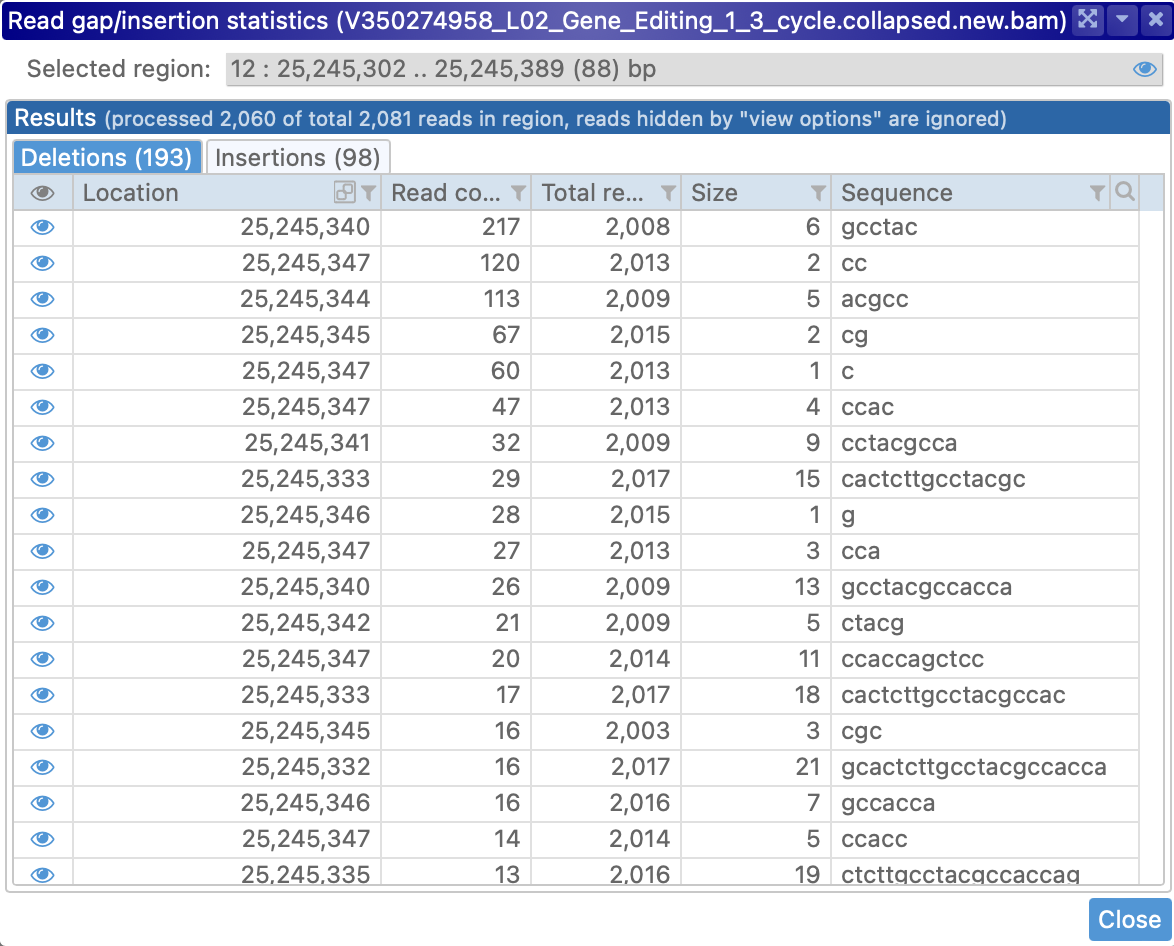

 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.