Create Function
The command create is used to generate new data based on the records in the database. One of the data types that can be created is functional annotation. We can use well characterized protein sequences, such as SwissProt, to assign the functional annotation to the proteins that show a good match. For each gene in a track we can find the best matching protein from SwissProt with defined function somewhat implying that the gene performs a similar function.
The command
create function <mapset>
will find matches between the proteins from an annotation track and SwissProt. (We decided to leave 'function' instead of 'functional_annotation' just to save some typing).
Note
Before running the command create function please make sure that you have installed diamond (install diamond) and SwissProt (install swissprot)
If there are more than one annotation track with CDS, the program will ask to choose a track for the analysis:
PS> create function "Rice-platinum standard/Os GJ-temp: IRGSP-1.0 (Nipponbare)" -t
Assigning functional annotation by match to well-characterized proteins from SwissProt
Testing ...
- SwissProt file is present: passed
- DIAMOND binaries found: passed
- Working on map set Rice-platinum standard/Os GJ-temp: IRGSP-1.0 (Nipponbare)
More than one annotation track is found for map set Rice-platinum standard/Os GJ-temp: IRGSP-1.0 (Nipponbare). Which track should be used?
[0]: MSU gene models
[1]: Gnomon gene models
[2]: FGENESH gene models
[3]: Gramene Gene models
[4]: CSHL Gene models
Enter [TrackNumber] to use?
As usual, the map set can be provided via MapSetId or its MapSetPath, such as in the example above. The auto-complete feature will save quite some typing.
The test mode (-t) will check for the presence of the binaries of diamond and the SwissProt index. The queries for diamond are the protein sequences for the annotation track in question. They are extracted from the corresponding BLAST index files and saved in a form of a FASTA file. When the test reports success, run the analysis, usually in verbose mode (-v).
Running diamond for one track usually takes about half a minute.
DIAMOND output saved at /HDD4Gb/Temp/_blast/2_MSU.func
First lines of the output:
ANNOT_ID DESCRIPTION
------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
35381 Rab GTPase-activating protein 22 OS=Arabidopsis thaliana [evalue:7.22E-31]
35382 Rab GTPase-activating protein 22 OS=Arabidopsis thaliana [evalue:4.3E-31]
35384 Monocopper oxidase-like protein SKU5 OS=Arabidopsis thaliana [evalue:4.01E-275]
35389 cAMP-regulated phosphoprotein 21 OS=Homo sapiens [evalue:8.92E-5]
35390 cAMP-regulated phosphoprotein 21 OS=Homo sapiens [evalue:7.75E-5]
35391 40S ribosomal protein S5 OS=Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) [evalue:2.04E-104]
35397 Sphingosine-1-phosphate lyase OS=Saccharomyces cerevisiae (strain ATCC 204508 / S288c) [evalue:1.88E-110]
35395 Sphingosine-1-phosphate lyase OS=Saccharomyces cerevisiae (strain ATCC 204508 / S288c) [evalue:2.32E-113]
35396 Sphingosine-1-phosphate lyase OS=Saccharomyces cerevisiae (strain ATCC 204508 / S288c) [evalue:1.88E-110]
35399 Retrovirus-related Pol polyprotein from transposon opus OS=Drosophila melanogaster [evalue:4.67E-35]
35400 Probable LRR receptor-like serine/threonine-protein kinase At1g12460 OS=Arabidopsis thaliana [evalue:5.05E-21]
35401 Putative pentatricopeptide repeat-containing protein At3g13770, mitochondrial OS=Arabidopsis thaliana [evalue:2.86E-134]
35402 Probable bifunctional methylthioribulose-1-phosphate dehydratase/enolase-phosphatase E1 OS=Oryza sativa subsp. japonica [evalue:3.73E-107]
35403 Snurportin-1 OS=Gallus gallus [evalue:4.08E-46]
35405 Polyadenylate-binding protein-interacting protein 10 OS=Arabidopsis thaliana [evalue:2.13E-6]
Display [N]ext 15 lines; [S]ave functional annotation; [ESC] - cancel:
Once diamond is done, the program will list the first 15 lines of the produced file that went through some processing. The raw output file of diamond is analyzed. In case a gene model has multiple hits, the best (by evalue) match is selected. If several matches have the same evalue, the program will pick the one with a longer description.
At this stage you can review the next 15 lines or save the result to the database. The matches will be stored as an annotation qualifier 'SwissProt match':
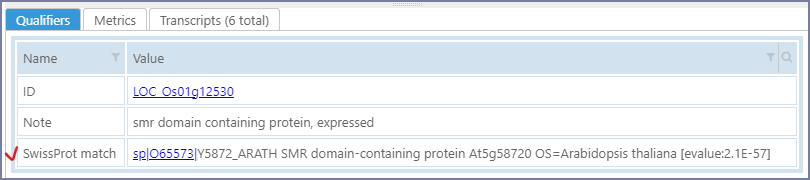
At the end of the processing, PersephoneShell will suggest to nominate this qualifier as containing the Function of a gene. This distinction is useful when searching for gene properties - the users can narrow down the search to searching in Function qualifiers only, thus reducing the chance for false positive hits. Internally, confirming the choice will add a new annotation_search entry. Please see the section for adding annotation_search for more details.
 Copyright © 2009-2024 by Persephone Software. All Rights Reserved.
Copyright © 2009-2024 by Persephone Software. All Rights Reserved.