Web Persephone: Exportable file formats
Every kind of Export dialog in Persephone provides a dropdown box containing a selection of output file formats, although the exact list of supported file formats depends on the type of item being exported. Click the Options button to configure the currently selected file format:

These options are described in more detail below.
CSV/TSV
A tab-separated (or comma-separated) list of items, with one item per line.

- Include Header: uncheck this to remove the column headers at the top of the file.
- Quote Values: by default, values are surrounded by quotes only if they contain special symbols (such as the currently selected separator). Check this box to quote all values instead.
- Separator: use this dropdown box to select the character that separates the values: tab, comma, or semicolon. The default is tab, since this separator is most often expected by other software, e.g. Microsoft Excel.
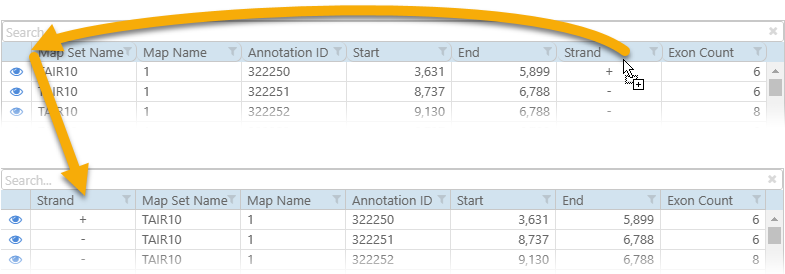
You can change the order of columns in the output file by drag-and-dropping them in the main preview table:
FASTA
A file containing sequence records, where each record is preceded by a header starting with ">". Before exporting a FASTA file, you must construct a valid header in the FASTA Options dialog (if the header is empty, the dialog will automatically open when you select FASTA as your output file format):

Configuring the header
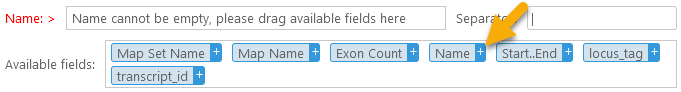
The Available fields box contains all of the columns that are selected in the column chooser. To add a column to the header, drag it into the Name box, or click the  button next to it:
button next to it:
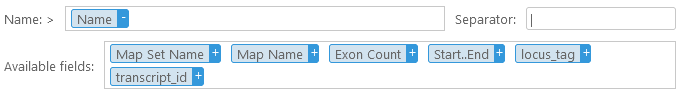
You can keep appending additional columns to the header; and you can drag a column out of the Name box (or click the  button) to remove it. You can also drag columns inside the Name box to reorder them. In addition, you can add an optional Prefix and/or an optional Suffix to the header; and you can also change the separator character (the default separator character is "|", as it is the one most commonly used by bioinformatics tools). Note that the separator is only inserted between individual fields in the header, but not after the prefix (or before the suffix). For example:
button) to remove it. You can also drag columns inside the Name box to reorder them. In addition, you can add an optional Prefix and/or an optional Suffix to the header; and you can also change the separator character (the default separator character is "|", as it is the one most commonly used by bioinformatics tools). Note that the separator is only inserted between individual fields in the header, but not after the prefix (or before the suffix). For example:
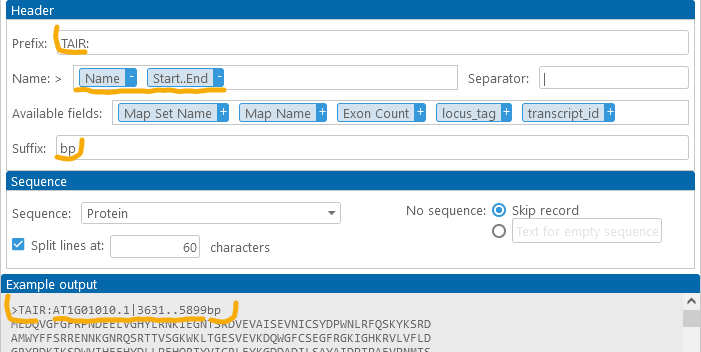
Configuring the sequence
The Sequence dropdown box contains all the sequence columns that are selected in the column chooser:

You can also select the option "Without sequence (FASTA headers only)" to omit the sequence; this can be useful if you only wish to export feature headers. For example:

By default, the sequences are split into 60-character-long columns, but you can choose any other line length (since the FASTA file format treats line breaks in the sequence as purely cosmetic). For example:

You can also uncheck the checkbox to display the entire sequence on one line.
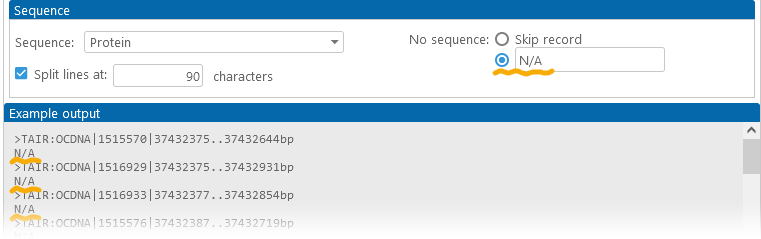
In addition, you can choose how to deal with missing sequences (for example, some annotations may not have protein sequences if they have no CDS coordinates loaded). By default, such records are completely omitted from the output file; but you can choose to include them, optionally replacing their sequences with placeholder text:
GENBANK
The GENBANK file format is only supported when exporting gene annotations.

You can choose to include or omit the sequence block (it is included by default). In addition, you can select which properties should be used for the /transcript_id and /gene fields. By default these are set to the group name of the annotation group and the individual name of the annotation, but you can instead select a particular qualifier:

Any Qualifiers that are selected in the column chooser will be included in the output file; in addition, the /translation field will be included if the Protein column is chosen (assuming the annotation has CDS). All other columns will be ignored.
GFF
The 9-column GFF file format is only supported when exporting gene annotations.

The source GFF field is always set to the annotation track's Method. The attributes field (last column) contains any additional columns that are selected in the column chooser (as well as the mandatory Annotation ID), although not all columns are supported:
- Map Properties: none.
- Basic Properties: only Name, Group name, and Annotation ID (which is always included).
- Sequence: none.
- Analysis: only Exon count, Protein complexity, and CDS length.
- Qualifiers: all selected qualifiers are included; however, qualifiers containing special characters (such as semicolons or newlines) are URL-encoded.
Note that you can rearrange the order of columns in the attributes field by drag-and-dropping them in the preview table.
BedGraph
The BedGraph format is only available for Quantitative tracks. It is a 4-column tab-separated list of blocks, where each block is associated with a numerical value:
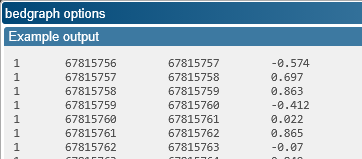
The columns are defined as follows:
- Map name
- Block start (typically in nucleotides, 1-based), inclusive
- Block end (typically in nucleotides, 1-based), exclusive
- Value (for the entire block)
Blocks defined in this way can not overlap, and will be in direct orientation (i.e. start <= end).
 Copyright © 2009-2025 by Persephone Software. All Rights Reserved.
Copyright © 2009-2025 by Persephone Software. All Rights Reserved.